

植物根系生长在土壤中,其周围和内部拥有丰富的微生物群落[1]。微生物菌群之间的动态平衡对植物的生长发育和抗逆性至关重要[2-4]。植物根际微生物菌群的种类和丰度受多方面影响:1)土壤中微生物的种类和数量[5-6];2)植物的基因型[7-8];3)环境因素,如温度、湿度、土壤板结度、土壤pH值等[9]。根系与根际微生物相互影响:根际微生物(有益菌/有害菌)影响根系生长,而根系通过分泌多种物质(如香豆素、硫代葡萄糖苷等)影响根际微生物的富集[10-12]。已有研究表明,根际微生物菌群可以改善植株根部对营养元素(如氮、磷、铁等)的吸收[7,13 -15]。在模式植物拟南芥中发现,控制内皮层功能的基因有助于植物菌群的装配;同时,微生物菌群驱动根系内胚层的分化,调控根系营养稳态[15]。
目前,植物对根际微生物菌群的调控机制还有待深入研究。已有研究揭示,植物通过调整根部代谢途径来调控根际微生物群落的组成和功能,从而提升其对逆境胁迫(如干旱胁迫、温度胁迫、营养胁迫和盐胁迫)的适应能力[16-21]。例如,在干旱环境下,植物通过改变根系分泌的代谢物质(如脱落酸、水杨酸和3-磷酸甘油等)而影响根际细菌群落的结构和功能[22]。这种通过改变分泌物调控根际微生物群落结构和功能的现象也在植物应对营养胁迫时有所体现。在氮匮乏条件下,豆科植物根部通过分泌大量的类黄酮以富集固氮菌[14,23];在磷匮乏条件下,植物根系富集促进植物吸收磷酸盐的功能菌[24]。这些现象表明,调整根际微生物群落的组成和功能是植物适应逆境的重要途径之一。
1 材料与方法
1.1 实验材料
1.1.1 植物材料
玉米野生型自交系材料B73为实验室自存材料,突变体材料nhx7-1由实验室通过EMS诱变B73花粉获得。
1.1.2 载体与菌株
DH5α大肠杆菌感受态购于北京擎科生物科技有限公司,pDONR207载体购于上海泽叶生物科技有限公司。
1.1.3 主要试剂与引物合成
十二烷基硫酸钠(SLS)、EDTA和庆大霉素(Gentamicin)均购于生工生物工程(上海)股份有限公司;Trizol试剂盒(Invitrogen,Carlsbad,CA,USA)和饱和酚(酚纯度≥99%,pH值≥7.8)购于北京索莱宝科技有限公司;NaCl、氯仿、异丙醇、乙醇均购于国药集团化学试剂有限公司;HiScript Ⅱ 1st Strand cDNA Synthesis Kit和ClonExpress Ⅱ One Step Cloning Kit购于南京诺唯赞生物科技股份有限公司;Oligo(dT)磁珠购于上海碧云天生物技术股份有限公司;Ribo-ZeroTM Magnetic Kit(Epicentre, Madison, WI, USA)购于北京中北林格科技发展有限公司;E.Z.N.A.soil DNA Kit(Omega Bio-Tek,Norcross,GA,USA)购于上海玉博生物科技有限公司。引物合成与测序均在北京擎科生物科技股份有限公司完成。微生物组测序由广东美格基因科技有限公司Illumina HiSeq2500测序平台完成。
1.1.4 主要仪器
PCR热循环仪(型号:ABF Gene Amp9700)购于美国ABI公司;琼脂糖凝胶电泳仪(型号:DYY-6D)购于北京六一生物科技有限公司;植物样品打样机(型号:TL2020)购于北京鼎昊源科技有限公司。
1.2 实验方法
1.2.1 植物材料种植与培养
玉米材料的种植分别在河南开封和海南三亚进行,在河南开封(北纬34°11'45″~35°01'20″,东经113°52'15″~115°15'42″)的播种时间为每年4月下旬,在海南三亚(北纬18°09'34″~18°37'27″,东经108°56'30″~109°48'28″)的播种时间为每年11月初。授粉结束后,对玉米地上植株和地下根系分别进行观察分析。
将玉米种子浸泡在去离子水中24 h后,置于萌发纸中进行萌发。6 d后收集根组织,用于RNA提取和转录组测序(每个样品3个重复)。同时,将玉米种子直接播种在装有土壤的盆钵中,生长30 d后收集根际土壤,并提取根际土壤中的微生物DNA,用于细菌16S rDNA和真菌ITS的测序分析。
1.2.2 DNA提取
取适量玉米叶片置于2 mL离心管中,加入2颗钢珠,用液氮速冻,然后置于打样机中磨样40 s。加入1 mL SLS提取液(100 mmol/L Tris-HCl,pH 8.0,1% SLS,20 mmol/L EDTA,100 mmol/L NaCl),震荡混匀。加入250 μL混合液(V饱和苯酚∶V氯仿∶V异戊醇=25∶24∶1),震荡混匀,静置5 min,12 000 r/min离心10 min。取上清液500 μL,转移至新的1.5 mL离心管中,加入等体积的异丙醇,混合均匀,12 000 r/min离心10 min,弃上清。加入1 mL 70%乙醇洗涤沉淀(重复1次),12 000 r/min离心5 min,弃乙醇,自然晾干。最后,加入适量的ddH2O以溶解DNA,保存于-20 ℃。
1.2.3 RNA提取及逆转录
按照说明书的要求,使用Trizol试剂盒提取玉米根组织的总RNA,使用Oligo(dT)磁珠从总RNA中纯化mRNA,并用Ribo-ZeroTM Magnetic Kit进行mRNA文库构建和Illumina HiSeq2500 PE-150系统测序。差异基因表达分析使用DESeq2软件进行,筛选标准为FDR<0.05和|log2FC|≥1。
1.2.4 测序载体构建
分别以野生型B73和nhx7-1突变体的cDNA为模板,用正向引物NHX7-107F(5'-ACAAAAAA-GCAGGCTCCATGGGCGGCGAGGCTGAG-3')与反向引物NHX7-107R(5'-TACAAGAAAGCTGGGTCCTACTGCTCCTGGGGCGGAG-3')进行PCR扩增,然后进行1%琼脂糖凝胶电泳,确认扩增产物的大小正确后,切胶回收PCR产物。利用ClonExpress Ⅱ One Step Cloning Kit通过重组反应将PCR片段与pDONR207载体连接,连接产物转化DH5α大肠杆菌,挑取单克隆进行菌落PCR验证。验证引物分别为正向引物NHX7-207F(5'-ACAAAAAAGCAGGCTCCATGGGCGGCGAG-GCTGAG-3')和反向引物NHX7-CXR(5'-GGTACGCTTAACAGATAATG-3')。验证正确后提取质粒,送公司测序,测序引物为通用引物pDONR-F(5'-CTGGCAGTTCCCTACTCTCG-3')和pDONR-R(5'-TGTAACATCAGAGATTTTGAGACAC-3'),测序完成并将序列拼接,再与NHX7(Zm00001d031232)基因的标准编码区序列(CDs)进行比对分析。
1.2.5 根际微生物基因组提取及测序
按照E.Z.N.A.soil DNA Kit的说明书要求,从0.5 g根际土壤样本中提取微生物群落基因组DNA。使用引物扩增细菌16S rDNA和真菌ITS基因的不同区域。使用PCR热循环仪,以引物338F(5'-ACTCCTACGGAGGCAGCAG-3')和806R(5'-GGACTACHVGGGTWTCTAAT-3')扩增细菌16S rDNA基因的超变区V3~V4;以引物ITS1F(5'-CTTGGTCATTTAGAGGAAGTAA-3')和ITS2R(5'-GCTGCGTTCTTCATCGATGC-3')扩增真菌ITS基因。测序在Illumina HiSeq2500平台完成。
1.3 数据处理
Alpha多样性(香浓指数和均匀度指数)、beta多样性(PCoA和NMDS分析)、NST群落构建和细菌相对丰度图利用Origin 2022绘制。KEGG富集气泡图、微生物Venn图以及根系形态与微生物-微生物多样性指标相关性图利用在线网站OmicShare Tools绘制。微生物共现网络图利用ChiPlot绘制。利用LSD(最小显著差异)统计学方法对数据进行差异显著性分析。
2 结果与分析
2.1 玉米材料基因型鉴定
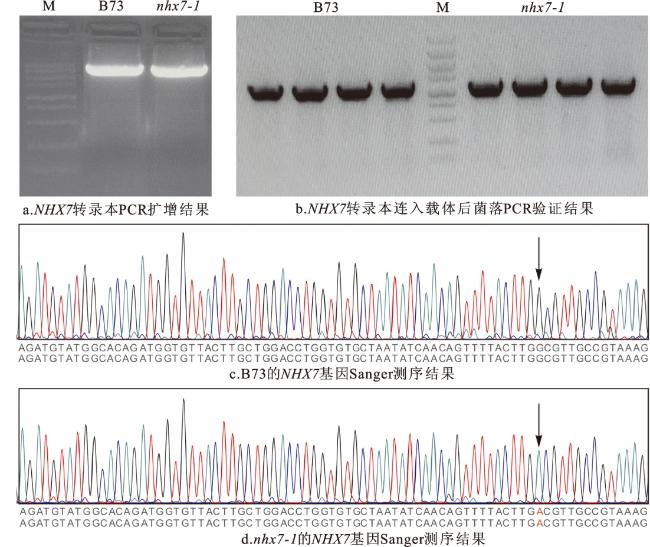
对野生型B73和nhx7-1突变体的基因进行扩增,连接于克隆载体pDONR207中,进行Sanger测序,结果如图1所示。测序结果显示,与B73植株的CDs序列相比,nhx7-1在NHX7基因CDs的第356个碱基位置由B73中的鸟嘌呤(G)突变成了腺嘌呤(A)。这种碱基替代导致NHX7蛋白的第119个甘氨酸(Gly/G)被替换成了天冬氨酸(Asp/D),说明nhx7-1为NHX7基因的非同义突变体。
2.2 NHX7突变导致玉米植株发育缺陷

2.3 NHX7突变改变根系代谢途径
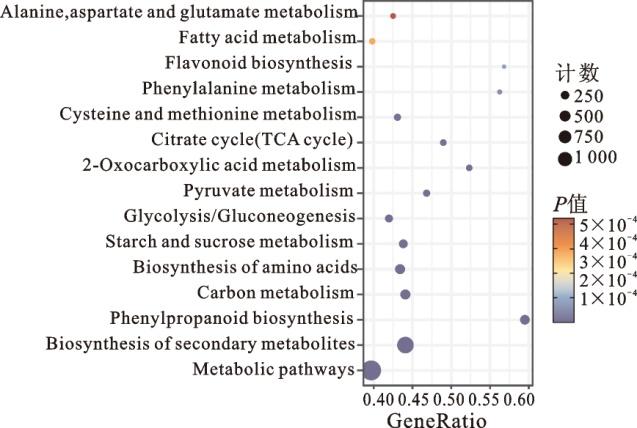
为解析NHX7可能参与的调控路径,对B73和nhx7-1的幼苗根系进行转录组测序,并对差异基因进行KEGG通路分析(图3)。结果显示,在B73和nhx7-1的差异基因中富集了与次生代谢物生物合成(biosynthesis of secondary metabolites),氨基酸生物合成(biosynthesis of amino acids),丙氨酸、天冬氨酸和谷氨酸代谢(alanine, aspartate and glutamate metabolism),苯丙氨酸代谢(phenylalanine metabolism),苯丙烷类生物合成(phenylpropanoid biosynthesis),类黄酮生物合成(flavonoid biosynthesis),2-氧代环戊烷羧酸甲脂代谢(2-oxocarboxylic acid metabolism),丙酮酸代谢(pyruvate metabolism),糖酵解/糖原异生(glycolysis/gluconeogenesis),淀粉和蔗糖代谢(starch and sucrose metabolism),三羧酸循环(TCA cycle),半胱氨酸和蛋氨酸代谢(cysteine and methionine metabolism),碳源代谢(carbon metabolism)以及脂肪酸代谢(fatty acid metabolism)等相关的通路。这些通路参与根系代谢物的合成代谢,说明NHX7基因突变可能导致根系代谢物含量的改变。
2.4 细菌与真菌相对丰度分析
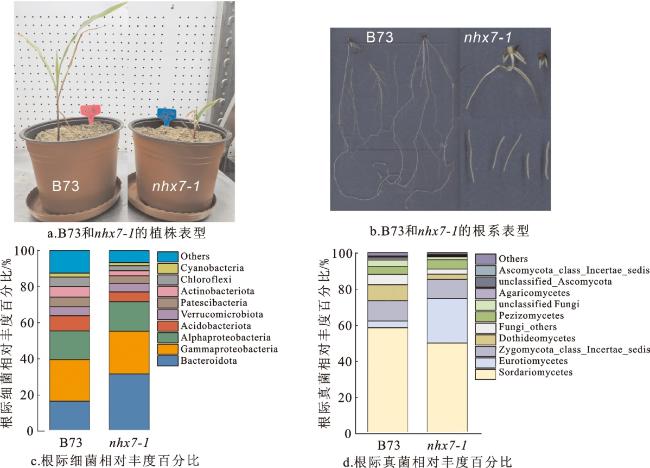
通过对根际土中的细菌16S rDNA进行测序,发现在nhx7-1突变体中,拟杆菌门(Bacteroidota,31.64%)、γ变形菌门(Gammaproteobacteria,23.66%)、α变形菌门(Alphaproteobacteri,16.36%)、酸杆菌门(Acidobacteriota,5.39%)、疣微菌门(Verrucomicrobiota, 4.76%)、浮游菌门(Patescibacteria, 4.19%)、放线菌门(Actinobacteriota, 2.8%)、绿弯菌门(Chloroflexi, 2.8%)、蓝细菌(Cyanobacteria, 1.62%)等细菌菌群显著富集(图4c)。以上在nhx7-1中显著富集的菌门在B73中的占比分别为:拟杆菌门(16.59%)、γ变形菌门(23.03%)、α变形菌门(15.84%)、酸杆菌门(8.44%)、疣微菌门(4.95%)、浮游菌门(5.36%)、放线菌门(5.76%)、绿弯菌门(5.29%)、蓝细菌(2.21%)。其中,拟杆菌门的丰度在nhx7-1中显著高于B73(图4c),表明B73和nhx7-1的拟杆菌门存在显著差异。
对根际土中的真菌ITS进行测序,发现B73与nhx7-1相对丰度前10位的根际微生物均为粪壳菌纲(Sordariomycetes)、散囊菌纲(Eurotiomycetes)、Zygomycota_class_Incertae_sedis、座囊菌纲(Dothideomycetes)、其他真菌(Fungi_others)、盘菌纲(Pezizomycetes)、未分类真菌(unclassified Fungi)、伞菌纲(Agaricomycetes)、未分类子囊菌门(unclassified_Ascomycota)、Ascomycota_class_Incertae_sedis。在B73中,粪壳菌纲为58.34%、散囊菌纲为3.73%、Zygomycota_class_Incertae_sedis为11.17%、座囊菌纲为8.93%、其他真菌为5.69%、盘菌纲为4.28%、未分类真菌为3.68%、伞菌纲为1.07%、未分类子囊菌门为0.92%、Ascomycota_class_Incertae_sedis为0.2%。在nhx7-1中,粪壳菌纲为49.86%、散囊菌纲为24.63%、Zygomycota_class_Incertae_sedis为10.59%、座囊菌纲为2.92%、其他真菌为2.83%、盘菌纲为5.15%、未分类真菌为1.61%、伞菌纲为0.35%、未分类子囊菌门为0.37%、Ascomycota_class_Incertae_sedis为0.67%。其中,座囊菌纲的丰度在B73中显著高于nhx7-1(图4d)。
2.5 根际微生物多样性分析
微生物群变异的总体模式通常是通过alpha和beta多样性来评估的。Alpha多样性是指特定区域或生态系统内的多样性,通常用该生态系统中的物种数量(即物种丰富度)来表示。如果需要研究生态系统之间物种多样性的变化,则需要测量beta多样性,它计算的是每个被比较的生态系统所独有的物种总数。
本研究分析了B73和nhx7-1根际微生物的alpha和beta多样性。在alpha多样性方面,B73根际细菌的香农指数(Shannon)和均匀度指数(pielou_evenness)显著高于nhx7-1,但B73和nhx7-1根际真菌的香农指数和均匀度指数差异不显著(图5a、b、c、d)。在beta多样性方面,主坐标分析(PCoA)和Anosim分析的结果显示,B73和nhx7-1根际细菌群落beta多样性差异显著(Anosim,r=0.337,P=0.023),非度量多维尺度(non-metric multidimensional scaling,NMDS)分析的stress值为0.031(<0.05),表明NMDS分析的结果具有较好的代表性(图5e、f);但二者的根际真菌群落beta多样性差异不显著(Anosim,r=0.058,P=0.311),NMDS的stress值为0.045(<0.05)(图5g、h)。上述结果表明,B73和nhx7-1根际细菌群落多样性差异显著,且B73根际细菌群落多样性明显高于nhx7-1,但二者在根际真菌群落多样性方面差异不显著。
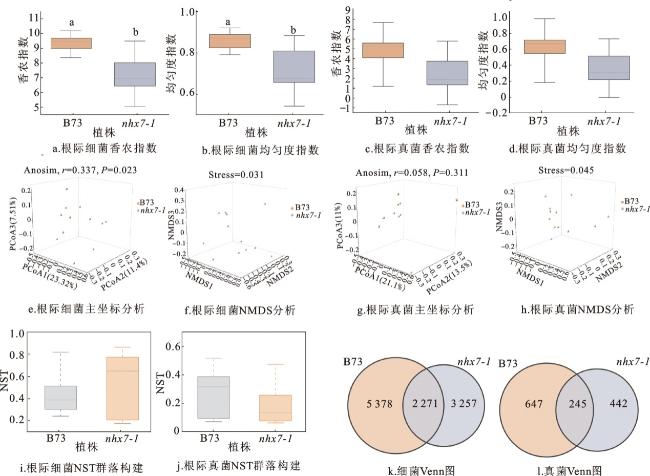
为解析B73和nhx7-1根际微生物群落的构建机制,采用标准化随机性比率(normalized shuffle test,NST)分析进行定量评估。NST(数值范围为0~1)量化了在群落构建中随机性的相对重要性,其值大于0.5表示群落构建更为随机,而小于0.5表示确定性过程影响更大。B73根际细菌的NST值在0.5以下,表明确定性过程对B73细菌群落的构建贡献较大,而nhx7-1根际细菌的NST值在0.5以上,说明随机性过程对nhx7-1细菌群落的构建贡献较大(图5i)。B73和nhx7-1的根际真菌NST值均在0.5以下,说明B73和nhx7-1的真菌群落构建过程主要由确定性过程主导(图5j)。
2.6 根系形态与根际微生物的相关性
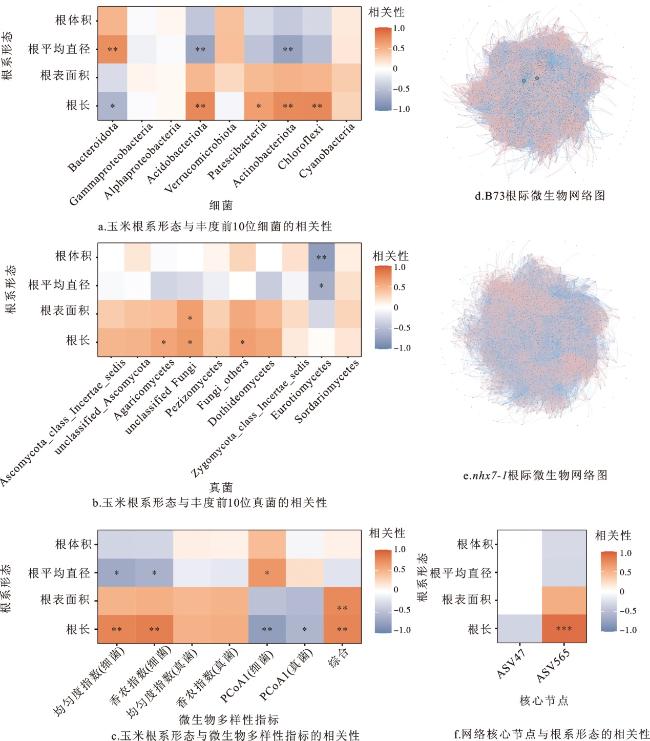
玉米B73和nhx7-1根系形态与丰度前10位的细菌相关性分析表明,根平均直径与拟杆菌门(Bacteroidota)显著正相关,与酸杆菌门(Acidobacteriota)和放线菌门(Actinobacteriota)显著负相关;根长与拟杆菌门(Bacteroidota)显著负相关,与酸杆菌门(Acidobacteriota)、浮游菌门(Patescibacteria)、放线菌门(Actinobacteriota)和绿弯菌门(Chloroflexi)显著正相关(图6a)。
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
玉米根系形态与丰度前10位的真菌相关性分析结果表明,根体积、根平均直径与散囊菌纲(Eurotiomycetes)显著负相关;根长与未分类真菌(unclassified Fungi)、其他真菌(Fungi_others)和伞菌纲(Agaricomycetes)显著正相关;根表面积与未分类真菌(unclassified Fungi)显著正相关(图6b)。
根系形态与微生物多样性指标的相关性分析结果显示,细菌香浓指数和均匀度指数与根长显著正相关,与根平均直径负相关。真菌香浓指数和均匀度指数与根形态没有相关性。细菌PCoA1与根长显著负相关,与根平均直径正相关;真菌PCoA1与根长负相关(图6c)。
核心节点与根形态的相关性分析表明,根系形态(根体积、根平均直径、根表面积、根长)与ASV47没有显著相关性,但根长与ASV565显著相关(图6f),说明ASV565对玉米根长具有重要影响,nhx7-1的根系缺陷可能与ASV565缺少有关,未来可着重研究核心节点ASV565在玉米根系生长中的作用。
3 讨论
3.1 微生物参与nhx7-1突变体根系生长的调控
3.2 根系分泌物影响根际微生物的富集
3.3 根际微生物调控根系生长
根际微生物种类丰富,其中许多微生物通过抑制病原体入侵和帮助植物从土壤中获取养分而使植物受益[14,43-44]。最近有研究报道,根际促生菌通过影响生长素信号通路、极性运输以及YUCs介导的生长素合成,调控拟南芥侧根发育[45]。根源性黄酮通过促进根际草酸杆菌科细菌的富集,促进玉米侧根发育突变体lrt1在氮剥夺条件下的根系发育和养分吸收[14]。本研究中根际微生物组测序结果表明,nhx7-1根际细菌和真菌的香农指数和均匀度指数均低于野生型,但仅根际细菌之间差异显著;2种植株根际细菌群落组成差异显著,而真菌群落组成差异不显著。根系形态与微生物相关性分析结果显示,根系形态受根际细菌群落的影响更大,其中核心节点ASV565与根长显著相关,推测ASV565可能是根系形态的主要影响因素。
4 结论
玉米野生型B73与nhx7-1突变体的根际细菌和真菌群落多样性组成展现出不同规律;在alpha多样性方面,nhx7-1根际细菌和真菌的香农指数和均匀度指数均低于B73,且根际细菌在B73和nhx7-1之间差异显著;在beta多样性方面,B73与nhx7-1根际细菌群落组成差异显著,真菌群落组成差异不显著。在群落构建方面,B73细菌群落构建为确定性过程,nhx7-1细菌群落构建为随机性过程;2种材料的根际真菌群落构建均为确定性过程。核心节点ASV565与根长显著正相关。