

药物开发过程通常包括4个主要阶段:药物发现、先导化合物设计、临床研究和批准上市[1]。整个过程通常耗费约12年的时间,需要近26亿美元的资金投入[2]。加快这一进程并降低成本将带来显著的经济和人力效益。然而,由于化学空间表现的离散性和巨大规模,早期类药物分子的优化任务极具挑战[3]。据估计,潜在类药物化学空间中分子的数量为1023~1060,其中仅成功合成了约108个分子[4]。如何有效探索未被发现的化学空间,并设计具备所需特性的先导化合物成为药物发现过程中至关重要的一步[5]。
传统的计算分子设计方法,如基于结构的[6]、基于配体的[7]和基于药物团的[8]方法需要丰富的领域知识和专家经验,且产生分子的质量和多样性也严重依赖所使用的化学文库和搜索策略,如遗传算法[9]或离散插值方法[10]。尽管高通量筛选[11]和虚拟筛选[12]这2种技术可以提升化学空间探索的速率,但它们受限于现有的分子文库,仅能对已有分子进行筛选,无法面对未知的化学空间;同时,高通量筛选伴随着耗时长、试错成本昂贵和使用门槛高等问题,这对于资源的有效利用和研发效率的提升构成了一定的挑战[13]。近年来,随着深度学习的不断发展,深度生成模型为分子设计提供了新的思路,如循环神经网络(recurrent neural network, RNN)[14]、变分自编码器(variational autoencoder, VAE)[15]、生成对抗网络(generative adversarial network, GAN)[16]和Transformer[17]。例如,Xu等[18]使用阈值区间和状态变化对log D和溶解度进行编码,旨在优化目标分子以满足某种特定的分子性质。Wang等[19]结合知识蒸馏和强化学习,使用条件Transformer学习结构-属性关系并生成满足多约束条件的新型分子。借鉴自然语言处理(natural language proessing,NLP)领域的方法,将分子视为一种特殊的“生物语言”并用一系列标记来表示,例如简化分子线输入系统(simplified molecular input line entry system,SMILES)[20]。
SMILES是一种简单的字符串线性表示方法,专门为化学领域中的计算机应用而设计[21]。它基于分子图理论的原理,并采用独特的规则对分子结构进行规范化,逐渐成为计算化学中的标准工具[22]。然而,在深度学习中对这种非连续的表示进行建模是一项颇具挑战性的任务[23]。此外,由于SMILES语法约束的存在,生成的大量字符串并不符合分子的有效性。据报道,这些深度生成模型的无效输出率为4%~89%,分子环未闭合是其中最常见的错误类型[24]。
分子环系统是药物化学中的关键骨架成分,在分子特性中发挥着重要作用[25-26]。一个关键的挑战在于,传统标准的自回归式语言生成模型是单向的[27]。在生成任务中,模型通常利用上一时刻的信息来预测下一时刻的生成,例如生成式预训练(generative pre-trained, GPT)[28]。然而,这却限制了模型对分子全局语义结构的理解。从本质上而言,SMILES字符串是分子图的一种线性表示方法,而分子图并不具备特定的起点和终点。SMILES可以从任意非氢原子开始,并向环的任意左右方向进行编码(见图1a)。这种非唯一性和非方向性同时也强调了双向约束的重要性。Xu等[29]将分子结构中的环系统简化为粗粒度节点,从而在生成分子树后再单独对其中的环节点进行实例化。虽然这在一定程度上提高了环结构的有效性,但这却破坏了生成时的上下文连贯性。为此,Wang等[30]通过将分子分解为有序的环与非环片段序列,在生成时使用门控循环单元(gated recurrent unit, GRU)依次解码成SMILES序列。但这种基于片段的增长方式却限制了生成模型的搜索空间。此外,他们不仅没有考虑到上述SMILES的双向性,还忽视了长期依赖关系中存在的信息遗忘风险[31]。这对于生成有效的分子序列也十分不利。
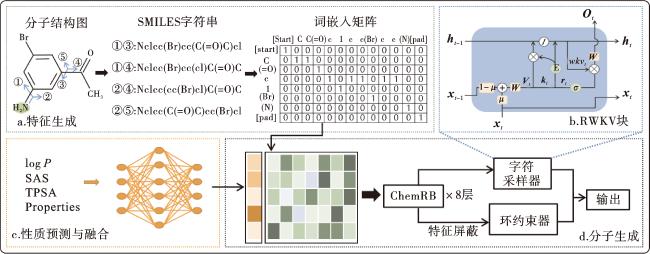
因此,为了克服上述限制,我们提出了Chemical RWKV BERT(ChemRB)方法,其框架如图1所示。与传统的单向方法相比,在ChemRB中非环字符生成是单向的,但分子环的形成是双向的。受BERT预训练[32]的启发,我们引入了“环级特征预测”任务来克服之前的单向性限制。通过随机屏蔽分子环元素,目标是根据被屏蔽词的上下文预测其原始词汇。此外,为了解决长依赖推理过程带来的信息遗忘风险,我们还提出了“全局跨度闭合预测”任务来联合预训练环约束器。值得注意的是,鉴于RWKV[33]在长序列文本建模上的优秀表现,我们首次引入该机制并应用于分子生成。为了实现这一点,我们基于所有训练的SMILES字符串构造了一个词汇表,用于将一条完整序列划分成单独的标记。此外,我们还引入了一个额外的自编码预测器来学习和预测药物分子的性质,并在EGFR抑制剂重新设计中证明了我们模型的实用性。
1 相关工作
1.1 分子有效性
表1 生成的闭合分子和未闭合分子Tab.1 Generated closed molecules and unclosed molecules |
| 类别 | 生成的分子 | 有效性 |
|---|---|---|
| Closed Ring | c1ccccc1 | 是 |
| Unclosed Ring | c1ccccc | 否 |
| Closed Ring | Nc1ccc2cncnc2c1 | 是 |
| Unclosed Ring | Nc1ccc2cncncc1 | 否 |
| Unclosed Rings | Nc1ccc2cncncc | 否 |
除了常见的基于原子的生成方式(即每步只有一个原子被添加到生成分子)外,基于片段的生成模型也可以在一定程度上提高生成的有效性[39],例如将官能团也作为分子结构的生成块。Jin等[40]利用连接树(joint tree,JT)将分子分解成诸多有效的子图并记录它们的相对顺序,然后训练模型基于有效子图及其相互作用来生成有效分子,从而避免了中间可能的无效体。Ishitani等[41]同样基于连接树分解算法得到有效的分子片段,之后通过长短期记忆网络(long short-term memory, LSTM)将连接树编码成隐变量并建立强化学习模型,最后逐步生成可逆连接树优化后的分子。Chan等[42]提出了基于多级自我对比学习的分层生成模型,用于改善偏差控制和数据效率。然而,由于基于片段模型的增长步长比基于原子模型的增长步长更大,这限制了生成模型的搜索空间。
1.2 RWKV机制
随着Transformer架构的提出,注意力机制在许多领域内都大放异彩[43]。然而注意力机制在计算和内存需求上具有二次复杂度,相对地,RNN在这方面表现出线性扩展,但会受到并行化和可扩展性的限制。
在传统注意力机制中,序列间不同位置嵌入的注意力分数可以表示为
$ A(q, k, v)_{t}=\frac{\sum_{i=1}^{T} v_{i} \mathrm{e}^{q_{t}^{T} k_{i}}}{\sum_{i=1}^{T} \mathrm{e}^{q_{i}^{T} k_{i}}}.$
为了解决注意力机制的二次复杂度问题,Zhai等[44]提出了无注意力Transformer(attention free Transformer,AFT式中记为AFT),其定义为
。
式中:w∈RT×T是一组可学习的成对位置偏差;T表示序列长度。通过假设序列间不同位置存在的绝对偏差w,对每个目标位置t执行加权平均,AFT成功避免了Q和K的点积自我关注并降低了内存复杂度。
受AFT启发,Peng等[33]设计了一种随时间衰减的位置偏差w(见公式3)。通过引入过去信息的接受向量r(见公式4),RWKV可以循环迭代地复用上一时刻的wkv分数(式中记为z),从而有效结合RNN的时间步递归特性和Transformer的全局注意力机制(见公式7和8)。其中,k(key)和v(value)类似于传统注意力中的K与V的向量(见公式5和6)。与传统RNN相比,RWKV能够在充分学习上下文信息的同时,有效处理和建模它们之间的长期依赖关系。这对于复杂且特殊的“SMILES语言”尤为重要。
2 方法
2.1 ChemRB模型
ChemRB模型的架构如图1所示。模块a负责提取分子特征。它将分子结构转化为SMILES字符串,并以独热编码的形式通过词嵌入矩阵将其转换为高维表示的特征向量,作为输入前的初始特征。模块b为RWKV的时间混合块。为了测试这种架构在药物发现领域的前瞻性应用,我们将其作为字符采样器的基本单元。在任意给定的第t个时间步长,网络由下面的一组方程描述:
$ \boldsymbol{w}_{t, i}=-(t-i) \cdot \boldsymbol{w}, i \in\{1,2, \cdots, t\} ;$
$ \boldsymbol{r}_{t}=\left[\left(1-\boldsymbol{u}_{r}\right) \boldsymbol{x}_{t-1}+\boldsymbol{u}_{r} \boldsymbol{x}_{t}\right] \cdot \boldsymbol{W}_{r} ;$
$ \boldsymbol{k}_{t}=\left[\left(1-\boldsymbol{u}_{k}\right) \boldsymbol{x}_{t-1}+\boldsymbol{u}_{k} \boldsymbol{x}_{t}\right] \cdot \boldsymbol{W}_{k} ;$
$ \boldsymbol{v}_{t}=\left[\left(1-\boldsymbol{u}_{v}\right) \boldsymbol{x}_{t-1}+\boldsymbol{u}_{v} \boldsymbol{x}_{t}\right] \cdot \boldsymbol{W}_{v} ;$
$\boldsymbol{o}_{t}=\left[\sigma\left(r_{t}\right) \odot \boldsymbol{z}_{t}\right] \cdot \boldsymbol{W}_{o \circ}$
式中:wt,i表示时刻i和t之间的位置偏差;u表示在当前时刻t对输入向量xt的关注度;k和v类似于传统注意力机制的key和value向量;W为不同模型中的权重矩阵。将模块a提取的分子初始特征xt顺序输入,随着时间t的增加,模型对[1,t]的位置区间进行加权求和得到wkv分数,然后乘以接受度σ(rt)得到新的输出ot。重点在于,当t+1时刻,网络仅需计算此刻对应的vt+1 和 ,至于2个复杂的累加计算则可以直接复用上一时刻的结果(公式(7)),从而使得RWKV达到类似RNN的自回归推理模式。因此,每次输出的结果都依赖于当前时刻的最新状态,从而在解码时可以很好地捕获长距离依赖性。
模块c为自编码预测器,由编码器、潜在空间和解码器组成。它从编码后的字符串特征中提取和重建分子信息,然后输出预测的分子属性并计算回归误差。通过学习潜在空间的属性向量表示,旨在引导和优化新药物分子的属性。模块d负责分子生成,由字符采样器和环约束器并行组成。字符采样器包括8个RWKV层,通过最小化交叉熵来减少预测字符与真实字符之间的概率误差;环约束器包括8个Transformer层,通过添加不同的输出层,分别对应于不同的预训练任务。因此,ChemRB模型的联合损失函数定义为
$L_{\mathrm{ChemRB}}=L_{\mathrm{Cs}}+L_{\mathrm{Ap}}+L_{\mathrm{Rc}}$
式中:LCs表示字符采样器预测的交叉熵损失;LAp表示自编码预测器的总误差;LRc表示环约束器两个预训练任务的总误差。这3种损失都是每批样品的平均值。
2.2 字符采样器
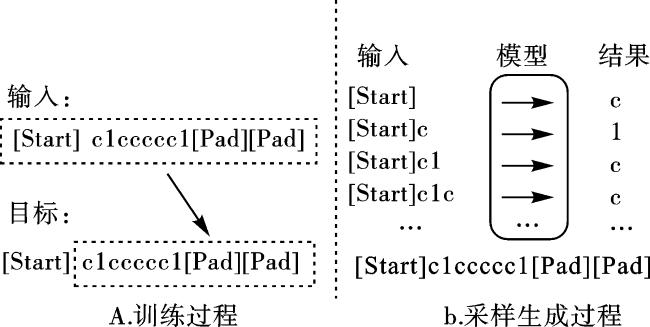
字符采样器的训练过程如图2a所示。由于分子序列长度各不相同,训练时每个分子被填充到最大SMILES字符串的长度n。将前n-1个字符作为输入,最后n-1个字符作为目标,那么训练损失LCs和Softmax函数P可以表示为
$L_{\mathrm{Cs}}=-\sum_{i=1}^{n-1} \boldsymbol{y}_{i} \log (\hat{\boldsymbol{y}}) ;$
。
式中:yt表示神经网络在时间步长t的输出向量; 对应于向量yt的第k个元素;K表示词汇表大小;T表示采样温度。在对下一个时间步骤t+1的SMILES字符xt+1采样后,可以构建一个新的输入向量xt+1,将其输入模型,通过yt+1和公式(11)得出P(xt+2|x1,x2,…,xt-1)。
采样生成过程如图2b所示。开始符号“[Start]”用于启动生成。在每一步采样中,最后一个采样的字符被作为生成序列中的下一个字符。继续采样,直到循环至设定的最大次数(我们将其设定为最大SMILES字符串的长度n)。“[Pad]”除了表示填充符号外,同时也代表着生成完整分子的完成。通过顺序依次提取“[Start]”和第一个“[Pad]”之间所有的SMILES 字符形成最终的生成结果。
2.3 自编码预测器
自编码预测器主要由编码器和解码器两个神经网络组成,具体结构如图3所示。编码器由三个堆叠的长短期记忆网络(long short-term memory, LSTM)层组成,负责将字符串分子的初始向量特征转换为连续低维的潜在空间表示。每层最终的隐藏状态向量拼接后,经由两个全连接层分别输出一组均值(μ)和标准差(σ)来描述对应的后验分布,即潜在空间的分布。假设编码器的输入为x,参数为θ。若Z为潜在变量,那么编码器分布就满足Qθ(Z/x)。解码器同样包含3个LSTM层,最后一层每个时间步的输出被映射回原始空间以重建分子样本。若解码器参数为Φ,同理满足分布PΦ(x/Z)。
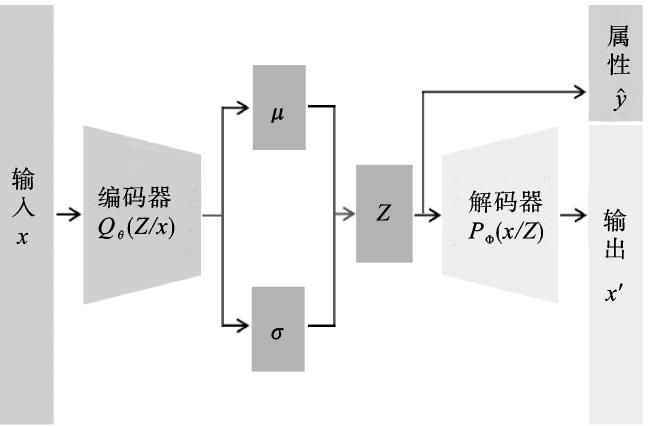
为了融合属性特征,我们还在解码器中引入一个全连接层用于分子性质预测。由于真实后验分布很难去计算,我们将其近似为标准高斯分布并用KL散度(Kullback-Leibler divergence)来衡量两个分布之间的差异。因此,自编码预测器的损失LAp为
$ \begin{aligned}L_{\mathrm{Ap}} & =K_{L}\left[Q_{\theta}(Z / x) \| P_{\Phi}(z)\right]- \\& E_{Z}\left[\log \left(P_{\Phi}(x / Z)\right)\right]+E_{\theta, \Phi}(y, \hat{y}) \end{aligned}$
$ E_{\theta, \Phi}(y, \hat{y})=E\left(y, P_{\Phi}\left(y \mid Q_{\theta}(Z / x)\right)\right) 。$
其中,KL[Qθ(Z/x)‖PΦ(z)]表示KL散度,-EZ[log(PΦ(x/Z))表示分子重建时的误差,Eθ,Φ(y, )表示性质预测的均方误差(MSE)。因此,分子的属性信息被强加于模型结构中,而不仅限于分子本身。
2.4 环约束器
不同于训练分子生成的单向语言模型,我们使用以下2个任务(Task1~2)来预训练环约束器,具体结构见图4。
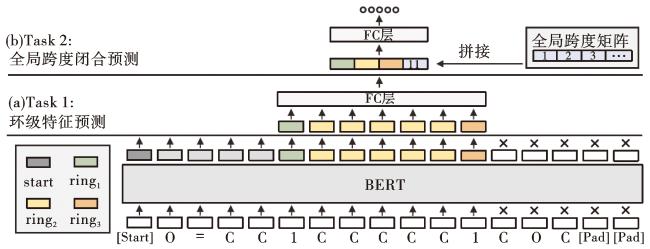
Task 1:环级特征预测。与BERT处理整条序列不同,我们的方法专注于对分子环元素进行掩码处理。随机选取其中的20%令牌位置,随后具体操作如下:1)80%的概率使用[MASK]标记;2)10%的概率随机替换为其他标记;3)10%的概率不做任何变化。随后,与掩码令牌相对应的最终隐藏向量通过第一个全连接层输出到词汇表上的Softmax中,预测每一个令牌的真实类别并计算交叉熵损失
$ \hat{\boldsymbol{y}}_{1}^{r}=\operatorname{Softmax}\left(\boldsymbol{w}_{1}^{r} \cdot \boldsymbol{x}^{r}+\boldsymbol{b}_{1}^{r}\right)$
值得注意的是,环约束器是为了优化分子环闭合以提高分子有效性,因此环元素之后的特征被视为噪声剔除。
Task 2:全局跨度闭合预测。在分子环约束这一下游任务中,随着分子环跨度的增大和分子环组成成分的复杂化,建模不同位置之间的相关性对模型来说变得越来越具有挑战性。为了解决这种长距离依赖建模中的信息遗忘风险,我们引入了全局跨度这一重要特征。全局跨度提供了分子环大小及其相对位置的准确信息,有助于模型更准确地捕捉开口标记和闭合标记之间的对应关系,从而提高预测精度。给定闭合标记在整个序列中的位置,我们从一个可学习的全局跨度矩阵中查找相应的嵌入表示gspan。在分别得到开启令牌和关闭令牌的深度双向表示r1和r3后,对中间特征进行平均池化,得到它们之间的关系r2。将这4种向量表示拼接(º)后,我们得到了最终的输入
$ \boldsymbol{x}^{g}=\boldsymbol{r}_{1} \circ \boldsymbol{r}_{2} \circ \boldsymbol{r}_{3} \circ \boldsymbol{g}$
输入xg经过第二个全连接层进行二分类预测:
$ \hat{\boldsymbol{y}}_{2}^{g}=\sigma\left(\boldsymbol{W}_{2}^{g} \cdot \boldsymbol{x}^{g}+\boldsymbol{b}_{2}^{g}\right) ;$
$ L_{\mathrm{Rc}}=L_{\mathrm{task} 1}+L_{\mathrm{task} 2}$
式中:σ表示Sigmoid;Ltask1表示分子环上的多分类交叉熵;Ltask2表示关系分类上的二元交叉熵。给定一个置信阈值α,任何关系预测得分≥α都被认为是适当的关闭时间;否则,不进行闭合约束。
具体的优化流程与逻辑如图5所示。首先输入开始字符,并检查此时分子环的状态。如果分子环处于未生成状态或已闭合状态,字符采样器预测所有待采样字符的概率分布,并进行随机采样。然后将新采样的字符添加到序列中,开始下一次迭代。如果分子环处于打开状态,则将序列传递至环约束器,以确定是否应该在此时关闭环。当采样轮数超过设定的最大值时,循环终止,并评估所有生成的分子。值得注意的是,对于分子环的开口我们没有做任何限制,由模型随机采样确定是否或何时发生。
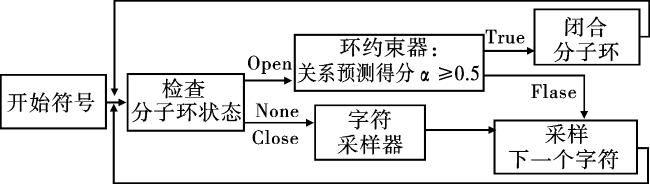
2.5 实验参数和评估指标
ChemRB模型使用Adam优化器训练10个迭代(epoch),学习率为6×10-4,批处理(batch)值为128。其中,编码模块的词嵌入(word embedding)为256;自编码预测器的LSTM隐藏层维度为128,潜在空间大小为256;字符采样器和环约束器分别由8层RWKV块和8层Transformer块堆叠而成,每层的上下文长度为最大分子长度n。最后一层的输出向量通过应用不同的全连接神经网络对应于不同的预训练任务,并计算总损失以反向传播更新参数。
有效性(validity,记作Val):生成有效分子的数量与生成总分子的数量之比。
$ V_{\mathrm{al}}=\frac{M_{\mathrm{valid}}}{M_{\text {generate }}}$
唯一性(uniqueness,记作Uni):有效分子中未重复分子的数量与有效分子的数量之比。
$ U_{\mathrm{ni}}=\frac{M_{\text {unique }}}{M_{\text {valid }}} 。$
新颖性(novelty,记作Nov):唯一分子集中未包含训练时的分子数量与唯一分子集的数量之比。
$ N_{\mathrm{ov}}=\frac{M_{\text {novel }}}{M_{\text {unique }}}$
新分子(new,记作New)/样本(sample,记作Sam):有效、唯一且新颖的分子数量与生成总分子的数量之比,表示模型生成全新分子的综合能力。
$ N_{\mathrm{ew}} / S_{\mathrm{am}}=V_{\mathrm{al}} \times U_{\mathrm{ni}} \times N_{\mathrm{ov} 。}$
3 结果与讨论
3.1 数据集
3.2 环约束器的性能结果
在生成模型中,温度超参数可以用于调整模型预测的概率分布,使得softmax输出更加平滑或集中(见公式11)。当温度越低时,模型输出的概率分布变得更尖锐集中,更倾向于选择概率较大的字符输出。这可能会导致生成结果的有效性提高,但多样性降低;当温度越高时,模型输出的概率分布变得更平滑随机,更有可能选择那些原本概率较低的字符输出。这可能会增加生成结果的多样性,但同时导致一些不合理的无效分子。因此,我们分别在不同温度下测试生成了10 000个分子,并检查了有效性等指标。结果如图6所示:随着采样温度的升高,生成的样本的有效性不断降低,唯一性和新颖性增加;综合评价指标“新分子/样本”以先增加后减少的趋势达到最佳。 在QM9数据集上的最佳性能为66.28%(采样温度为1.25),在ZINC数据集上的最佳性能为98.74%(采样温度为0.75)。
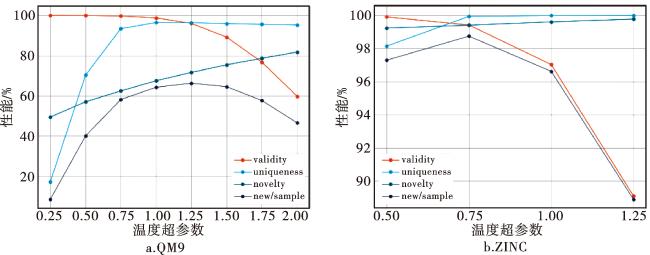
为了全面评价环约束器在ChemRB上的性能,我们比较了优化前后生成器在不同采样温度下的性能,如表2所示。可以看出,经过分子环约束后,有效性得到了显著提高,同时保持了较高的唯一性和新颖性。这表明环约束器充分学习了训练数据中分子环特征之间的关系,而并不局限于简单复制。相应地,新分子/样本比率的增加也验证了这一点。
表2 环约束器在基准数据集上的性能Tab.2 Performance of ring constrainer on the benchmark datasets |
| 数据集 | 温度超参数 | 有效性/% | 唯一性/% | 新颖性/% | (新分子/样本)/% |
|---|---|---|---|---|---|
| QM9 | 0.25 | 100(+0.01) | 17.16(-0.01) | 49.45(+0.01) | 8.49(-) |
| 0.50 | 99.96(+0.02) | 70.28(+0.01) | 57.03(-) | 40.06(+0.01) | |
| 0.75 | 99.80(+0.12) | 93.43(+0.01) | 62.48(+0.03) | 58.26(+0.10) | |
| 1.00 | 99.10(+0.35) | 96.44(+0.02) | 67.56(+0.06) | 64.57(+0.30) | |
| 1.25 | 96.52(+0.50) | 96.39(+0.01) | 71.62(-) | 66.63(+0.35) | |
| 1.50 | 89.86(+0.66) | 95.91(-) | 75.45(-) | 65.03(+0.48) | |
| 1.75 | 78.03(+1.29) | 95.65(+0.11) | 78.78(+0.13) | 58.80(+1.13) | |
| 2.00 | 61.64(+1.85) | 95.52(+0.26) | 82.22(+0.44) | 48.41(+1.83) | |
| ZINC | 0.25 | 100(+0.01) | 49.19(+0.01) | 99.20(-0.01) | 48.80(+0.01) |
| 0.50 | 99.93(+0.03) | 98.18(+0.04) | 99.21(-0.02) | 97.34(+0.05) | |
| 0.75 | 99.52(+0.11) | 99.96(+0.03) | 99.38(-0.02) | 98.86(+0.12) | |
| 1.00 | 97.50(+0.47) | 100(+0.02) | 99.63(+0.03) | 97.14(+0.52) | |
| 1.25 | 90.24(+1.13) | 99.99(-) | 99.78(+0.01) | 90.03(+1.14) | |
| 1.50 | 73.37(+2.02) | 99.99(+0.01) | 99.85(-) | 73.25(+2.02) | |
| 1.75 | 48.26(+2.17) | 99.93(+0.02) | 99.94(+0.04) | 48.20(+2.19) | |
| 2.00 | 24.59(+2.28) | 99.84(-0.01) | 99.96(+0.04) | 24.54(+2.28) |
注:括号中“+”表示优化后的绝对提高,“-”表示降低或持平,结果为10次平均后的百分比。 |
为了进一步了解未闭合环的优化率,我们收集整理了所有生成结果中的无效分子并进行分析。结果如表3所示,在QM9中,ChemRB生成器的无效率为3.98%,其中未闭合环占所有样本的1.84%;经过环约束器优化后,未闭合率降低至0.45%,优化率为75.54%。同理,在ZINC中,未闭合率由0.14%降低至0.03%,优化率为78.57%。这些结果表明环约束器在处理分子环无效性方面取得了显著的优化效果。
表3 环约束器在ChemRB上的优化率 单位:%Tab.3 Optimization rate of the ring constraint on ChemRB |
| 数据集 | 方法 | 无效率 | 未闭合率 | 优化率 |
|---|---|---|---|---|
| QM9 | ChemRB-WRC | 3.98 | 1.84 | 75.54 |
| ChemRB | 3.48 | 0.45 | ||
| ZINE | ChemRB-WRC | 0.59 | 0.14 | 78.57 |
| ChemRB | 0.48 | 0.03 |
注:ChemRB-WRC不包含环约束器。 |
3.3 显著性测试结果
为了量化分析环约束器优化前后的有效性是否存在显著性差异,我们对其进行了统计测试。具体而言,我们使用Shapiro-Wilk检验两组数据的正态性,并用Levene检验方差齐性(当P>0.05时,接受原假设H0:样本来自正态分布、样本方差齐性)。当样本符合正态分布且方差齐性时,我们进行独立样本t检验;当符合正态分布但方差非齐性时,我们进行Welch t检验;否则我们进行Mann-Whitney u检验(当P<0.05时,拒绝原假设H0:样本间没有显著差异,接受备择假设H1:样本间存在显著差异)。结果如表4所示。
表4 有效性统计测试Tab.4 Validity statistical test |
| 温度参数 | QM9 | ZINC | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| SW-优化前 | SW-优化后 | Levene检验 | 显著性测试 | SW-优化前 | SW-优化后 | Levene检验 | 显著性测试 | |||||||||
| 0.25 | 0.204 | 0.305 | 0.882 | 0.000 | 0.790 | 0.884 | 0.597 | 0.000 | ||||||||
| 0.50 | 0.092 | 0.406 | 0.279 | 0.002 | 0.584 | 0.692 | 0.365 | 0.001 | ||||||||
| 0.75 | 0.410 | 0.552 | 0.207 | 0.001 | 0.793 | 0.871 | 0.624 | 0.002 | ||||||||
| 1.00 | 0.172 | 0.174 | 0.385 | 0.002 | 0.684 | 0.037 | 0.702 | 0.014 | ||||||||
| 1.25 | 0.728 | 0.547 | 0.542 | 0.005 | 0.292 | 0.386 | 0.677 | 0.031 | ||||||||
| 1.50 | 0.034 | 0.407 | 0.168 | 0.017 | 0.093 | 0.127 | 0.048 | 0.016 | ||||||||
| 1.75 | 0.321 | 0.214 | 0.289 | 0.023 | 0.047 | 0.885 | 0.401 | 0.031 | ||||||||
| 2.00 | 0.760 | 0.045 | 0.750 | 0.026 | 0.158 | 0.046 | 0.676 | 0.045 | ||||||||
3.4 自编码预测器的优化结果
将融合了分子性质的潜在向量与字符串的特征向量拼接,作为新的输入。模型通过解析这些性质与分子结构之间的相关性,从而在生成时优化具有特定性质的药物分子。具体结果如图7所示,可以看出,分子属性的分布准确地以目标值为中心。即使在概率密度分布较低的区间下,也能明显以优于训练时的概率去生成特定目标的分子,如SAS=5和TPSA=100。在多条件优化时,可以看出8个不同的组合条件下生成的分子被明显区分为8个团簇,三维散点图很直观地展示了这一点。
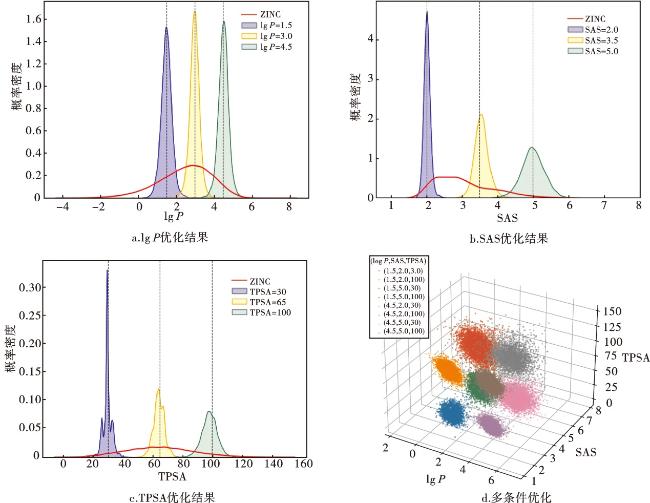
对于每个属性,我们计算了预测分布与真实分布之间的平均绝对误差(mean absolute error,MAE)、Pearson相关性和R2,结果如表5所示。
表5 自编码预测器的性质预测和优化能力Tab.5 Property prediction and optimization capability of autoencoding predictor |
| 数据集 | 分子性质 | 有效性 | 唯一性 | 新颖性 | 平均绝对误差 | Pearson相关性 | R2 |
|---|---|---|---|---|---|---|---|
| ZINC | lg P | 0.959 | 0.999 | 0.994 | 0.214 | 0.974 | 0.945 |
| SAS | 0.945 | 0.998 | 0.995 | 0.169 | 0.977 | 0.954 | |
| TPSA | 0.957 | 0.998 | 0.994 | 3.167 | 0.989 | 0.976 | |
| lg P-SAS-TPSA | 0.814 | 0.923 | 0.998 | 1.822 | 0.986 | 0.966 |
3.5 对比实验结果
一个优秀的生成模型不仅应关注有效性,还应致力于发现新的、具有潜力的化合物,从而推动化学领域的创新和发展。为了表明ChemRB的独特优势和先进性,我们将其与cRNN[14]、GrammarVAE[15]、MolGAN[16]、MolGPT[17]、FraHMT[30]、ARAE[53]、MGM[54]、GraphVAE[55]、GeoBFN[56]和BiCEV[57]等现有技术进行了对比。在每个基准数据集训练结束后,我们从训练好的模型中统一采样生成10 000个分子进行评估。每个数据集上温度为1.0时的模型性能如表6和表7所示。实验结果表明,ChemRB在新分子/样本这一综合指标上取得了最佳分数,优于多种先进的分子生成模型。这表明我们的模型有效地学习了分子的化学规律和结构特征。
表6 ChemRB和基准模型在QM9数据集上的性能Tab.6 Performance of ChemRB and benchmark models on QM9 dataset |
| 模型方法 | 有效性 | 唯一性 | 新颖性 | 新分子/样本 |
|---|---|---|---|---|
| MGM | 0.886 | 0.978 | 0.518 | 0.449 |
| GrammarVAE | 0.602 | 0.093 | 0.809 | 0.045 |
| GraphVAE | 0.557 | 0.670 | 0.616 | 0.261 |
| MolGAN | 0.981 | 0.104 | 0.942 | 0.096 |
| ARAE | 0.862 | 0.935 | 0.371 | 0.299 |
| MolGPT | 0.986 | 0.948 | 0.640 | 0.598 |
| GeoBFN | 0.969 | 0.924 | 0.653 | 0.585 |
| ChemRB | 0.991 | 0.964 | 0.676 | 0.646 |
注:加粗表明最优。 |
表7 ChemRB和基准模型在ZINC数据集上的性能Tab.7 Performance of ChemRB and benchmark models on ZINC dataset |
| 模型方法 | 有效性 | 唯一性 | 新颖性 | 新分子/样本 |
|---|---|---|---|---|
| cRNN | 0.784 | 0.899 | 0.452 | 0.319 |
| GrammarVAE | 0.310 | 1.000 | 0.108 | 0.033 |
| GraphVAE | 0.140 | 1.000 | 0.316 | 0.044 |
| ARAE | 0.903 | 1.000 | 1.000 | 0.903 |
| MolGPT | 0.962 | 1.000 | 0.995 | 0.957 |
| BiCEV | 0.958 | 0.957 | 1.000 | 0.917 |
| FraHMT | 1.000 | 1.000 | 0.968 | 0.968 |
| ChemRB | 0.975 | 1.000 | 0.996 | 0.971 |
注:加粗表明最优。 |
3.6 讨论
3.6.1 预训练和环表征
由于药物数据集的稀缺性,我们接下来将探索预训练可以为药物分子的生成带来什么。本质上,分子生成是一个表征学习过程,模型的性能取决于它从给定的初始信息中提取相关任务特征的能力。之前的结果已经证实了,与其他先进方法相比ChemRB所具有的优秀生成能力,突出了其捕获分子间复杂语法和语义关系的能力。在这里,为了测试预训练能否显著增强ChemRB提取分子特征的能力,我们分别用源数据集ChEMBL和目标数据集EGFR对ChemRB-WRC以及环约束器的各种预训练变体进行训练,结果如表8所示。实验表明,预训练可以有效增强生成模型对化学空间的探索能力,同时也进一步提高了环约束器的优化能力。
表8 不同预训练的ChemRB变体在两个数据集上的性能Tab.8 Performance of different pre-trained variants of ChemRB on two datasets |
| 变体 | ChemRB-WRC | 环约束器 | 有效性 | 唯一性 | 新颖性 | 新分子/样本 |
|---|---|---|---|---|---|---|
| 变体1 | E | 0.309 5 | 0.499 4 | 0.864 3 | 0.133 6 | |
| 变体2 | E | E | 0.316 6 | 0.514 9 | 0.876 3 | 0.142 9 |
| 变体3 | E | C+E | 0.367 4 | 0.539 5 | 0.889 8 | 0.176 4 |
| 变体4 | C+E | 0.718 4 | 0.945 9 | 0.948 6 | 0.644 6 | |
| 变体5 | C+E | C+E | 0.736 8 | 0.946 3 | 0.952 0 | 0.663 8 |
注:ChemRB-WRC不包含环约束器,C表示源数据集ChEMBL,E为目标数据集EGFR。 |
3.6.2 EGFR抑制剂的重设计
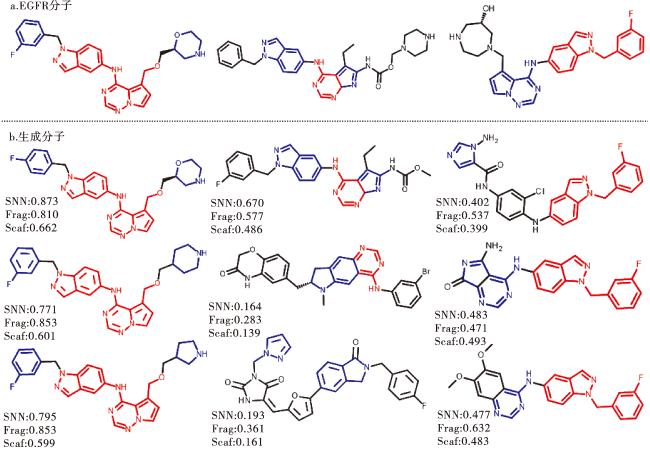
为了设计新的选择性EGFR抑制剂,我们选择预训练的变体5模型(表8)。在生成过程中,我们以lg P=2.5、SAS=2.0和TPSA=60为控制条件,目的是生成符合Lipinski's Rule of Five[51]的分子。通过运行该模型,我们采样生成了10 000个分子。除去无效、重复和训练集中的分子后,总共得到了2 487个分子。为了验证生成的分子是否具有与EGFR抑制剂相匹配的特性,我们使用RDKit计算了多个分子性质,包括lg P、SAS、TPSA、分子量、分子类药性、BertzCT、H-供体、H-受体和可旋转键的数量。然后,我们使用t-SNE算法对这些性质进行了2D降维并可视化,结果如图8所示。两组几乎完全重叠,表明生成的分子很好地再现了EGFR抑制剂的特性。
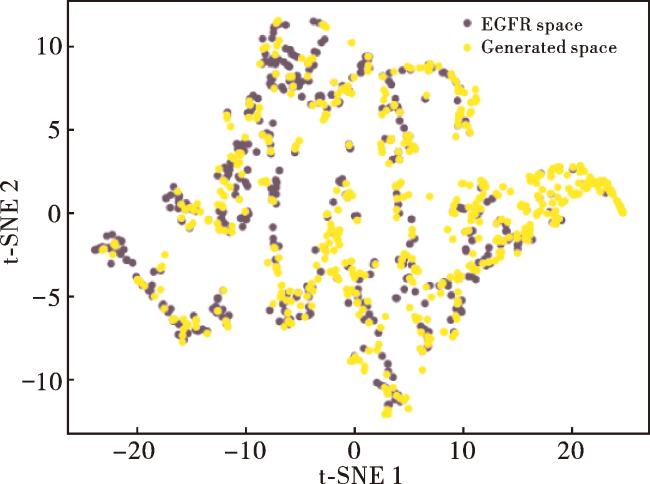
表9 两组分子集间的对接亲和力和相似性Tab.9 Docking affinity and similarity between two sets |
| 分子集 | 对接亲和力(PDB 2RGP) | 相似性 | |||||||
|---|---|---|---|---|---|---|---|---|---|
| (-∞,-7) | (-7,-5) | (-5,-4.25) | SNN | Fragment | Scaffold | ||||
| EGFR | 99.08% | 0.92% | |||||||
| Generated | 90.94% | 8.26% | 0.30% | 0.622 | 0.987 | 0.277 | |||
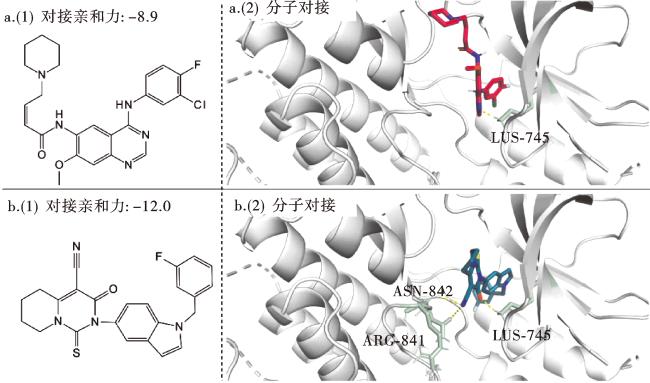
其中,对接亲和力越低,结合强度越强,分子配体靶向蛋白质的概率越高[59]。对接亲和值低于-4.25 kcal/mol表示有一定的结合活性,低于-5.0 kcal/mol表示结合活性良好,低于-7.0 kcal/mol表示结合活性极强。结果显示:542个EGFR抑制剂中,对接活性极强和良好的占比分别达到了99.08%和0.92%;而生成的2 487个分子集合中,对接活性极强、良好和一定的占比分别达到了90.94%、8.26%和0.30%,表明生成的分子与受体结构之间具有非常出色的相互作用。为了更直观地展示方法的有效性,我们挑选了部分EGFR分子和部分生成分子并对子结构进行着色处理(红色表示相同的子结构,蓝色则表示近似的子结构)。图9结果显示,模型不仅有效地生成了新的分子骨架,同时也成功学习了EGFR抑制剂分子的子结构。较高的SNN和Fragment值以及相对较低的Scaffold值也表明了这一点。
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
4 结论
本文提出了一个新的受双向分子环约束的生成模型ChemRB。与传统的单向编码自回归生成模型不同,我们通过引入两个预训练任务,成功地将分子环的生成扩展到深度双向架构。作为首次在这方面的探索和尝试,ChemRB对于提高生成分子有效性具有重要的借鉴意义,并可作为灵活扩展的应用方法。通过对温度超参数的详细考察,我们全面论证了环约束器增强有效性的能力,展示了其对无效分子环的优化能力。这一成就的意义在于,我们证明了在不牺牲唯一性和新颖性性能的情况下提高有效性是可行的。此外,我们构建了一个自编码预测器来重建和预测药物分子性质,指导生成器在生成过程中进行逆向解析并设计具有特定属性的药物分子。我们观察到,即使训练数据的性质分布较为均匀和平缓,生成时仍能以远超其概率密度集中优化分子性质。较低的MAE和较高的Pearson、R2值也证明了这一点。最后,我们还测试了模型在EGFR抑制剂上的实际应用表现。结果表明,我们可以在满足相同性质和相似度的情况下,成功地生成具有较高亲和力的新候选化合物。在未来的工作中,除了引入更多无效类型的处理外,还可以扩展ChemRB以集成日益复杂的数据,例如蛋白质组学和空间结构信息,为加速药物发现和分子设计提供有力指导。