

蛋白质和小分子配体之间的相互作用涉及多种生物过程,包括酶催化和信号转导。此外,药物通常充当配体与靶蛋白结合,发挥它们在药物发现中的作用。蛋白质-配体结合亲和力表示蛋白质与配体的相互作用强度,相互作用越强,配体靶向蛋白质的概率越高。因此,药物发现的关键目标之一是寻找与靶蛋白高度结合的候选配体[1-2],这些候选配体可以进一步开发成先导化合物。鉴定先导化合物的实验方法往往耗费大量的时间和经济成本。然而,蛋白质-配体相互作用(PLI)的计算预测可以显著减少需要的资源、时间和成本,减少筛选新治疗药物需要的物理实验。可靠的PLI预测方法可以有助于从众多候选药物中优先选择合适的候选药物进行实验测试,从而加快药物设计过程,有效指导药物化合物的开发[3-4]。
表面等离子体共振(SPR)和等温滴定量热法(ITC)在有辅助实验的情况下能有效测量蛋白质-配体结合亲和力[5⇓⇓-8],但测量结合亲和力的过程复杂,费用昂贵。基于经验的传统评分函数很大程度上依赖人工设计的特征和复杂的计算,难以准确预测蛋白质-配体的结合亲和力,例如X-Score、ITScore和量子力学/分子力学(QM/MM)评分函数。还有一些方法使用分子力学泊松-玻尔兹曼、表面积连续溶剂化方法(MMPBSA)和Helms等人的方法在搜索对接姿势后估计结合亲和力的近似值[9-10],但是准确性仍然不高;MMPBSA方法[11-12]时间复杂度高,计算结果不准确;Helms等的方法计算结合亲和力准确,但需要极大的计算资源,也不适用于大规模的虚拟筛选。
1 相关研究
近年来基于机器学习(ML)的方法已被应用于预测结合亲和力,如RF-scores[13]和NNScore[14]。RF-scores是一种使用分子间相互作用特征构建的随机森林回归模型。NNScore是一种模拟大脑微观组织的神经网络评分函数。这两种方法已被用于虚拟筛选中重新评分对接结果和发现先导化合物[15⇓-17]。传统的ML方法预测结合亲和力的性能很大程度上取决于蛋白质-配体的表达方式,如蛋白质-配体拓扑指纹[18⇓-20]。虚拟筛选方法中,使用对接软件分析输出,生成或人工提取蛋白质-配体相互作用的特征,过程费力又复杂,传统ML方法不能有效地处理大规模数据。深度卷积神经网络(DCNN)不依赖专家进行特征选择[21-22],原始数据的非线性变换揭示数据隐藏的信息,这使得DCNN比传统ML方法更适用于药物发现、虚拟筛选和结合亲和力的预测。例如AtomNet和 都应用了DCNN模型,以配体为中心的矢量化立方体网格作为蛋白质-配体复合物的特征,能较好地预测蛋白质-配体的结合亲和力。深度学习(DL)技术能最大限度减少特征提取的时间。然而,深度学习方法存在黑箱决策问题,Lim团队提出采用注意力机制有助于解决深度学习的黑箱决策[24]。
本文提出一种基于LSTM模块和注意力机制的深度学习模型(DLLSA)预测蛋白质-配体结合亲和力。DLLSA由嵌入LSTM模块与Spatial-Attention模块的网络块以及全连接块组成。LSTM模块能解决传统编码器-编码器结构在编解码时依赖内部固定长度向量的问题,可以获取特征数据长距离之间的隐藏信息;Spatial-Attention模块有助于发现关键的局部结合信息,抑制无效信息,增强网络的非线性表达能力。两个模块从蛋白质-配体接触特征中获取非局部与局部表征,处理后的特征传递到全连接网络块。实验采用Pearson相关系数(R)、一致性指数(CI,记作IC)、均方根误差(RMSE,记作ERMS)和标准差(SD,记作DS)作为评价指标。CASF[25-26]基准集的最终预测结果表明DLLSA模型达到了较好的精度。
2 实验数据与方法
2.1 实验数据
实验数据集来源于PDBbind数据库[27],使用PDBbind(v.2020)版本,主要分为通用集、精炼集和基准集。通用集、精炼集、基准集CASF-2013与基准集CASF-2016分别提供了19 443、5 316、195和285个蛋白质-配体复合物。为确保模型的正常运行,我们对数据进行了筛选并去除数据之间的冗余数据,最终确定基准集CASF-2013的161个复合物和CASF-2016的254个复合物作为测试集,通用集和精炼集消除基准集冗余的数据,去冗余后,通用集中的12 000个复合物作为训练集,精炼集中的2 827个复合物作为验证集。对于蛋白质-配体复合物的每种结构,相应的结合亲和力由解离常数(Kd)、抑制常数(Ki)或半抑制浓度(IC50)的负对数(pKa)表示。
2.2 实验方法
2.2.1 指纹特征提取
模型中构建分子指纹(FP)作为分子结构表征。分子指纹使用固定长度的布尔值或整数向量表示分子。本模型定义指纹特征为距离“壳”中特定原子(蛋白质)-原子(配体)的组合。配体中原子与蛋白质中原子的最小距离作为配体与蛋白质的距离。围绕最小距离中的配体原子,定义N层连续堆积的“壳”。“壳”是边界K-1和K之间的空间,例如第n层“壳”(SN)是边界K=n-1与K=n,1≤n≤N之间的空间。第一个“壳”的半径为d0,其他“壳”的厚度为 δ。“壳”的示意图如图1所示。
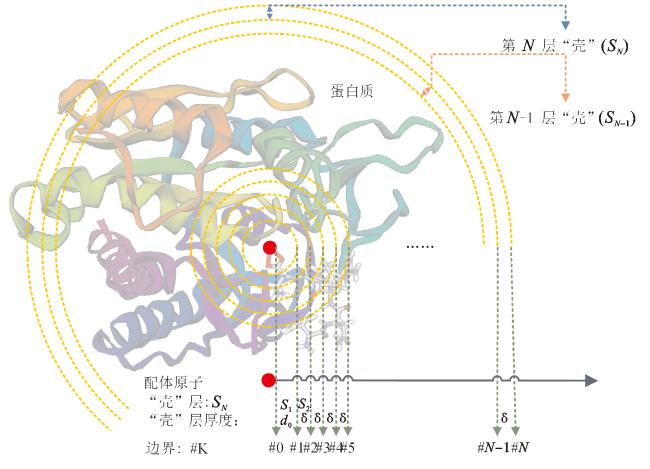
如图1所示,#K是以配体中原子为中心的“壳”的边界,第一个“壳”是边界#0和边界#1之间的空间。每层“壳”的定义如下,其中Ki是第i层“壳”的边界:
蛋白质中原子(EP)和配体中原子(EL)定义为碳(C)、氢(H)、氧(O)、氮(N)、磷(P)、硫(S)、卤素(Helogen)和剩余元素(ARE),8种元素用来量化原子之间的接触。Halogen代表卤素元素氟(F)、氯(Cl)、溴(Br)、碘(I)中任意一种,ARE代表所有剩余元素。为了保证模型的泛化能力,保留P、Halogen和ARE这些在蛋白质中存在量较少的元素。
= Cr,l, Ts∈EP,Tt∈EL,
Cr,l=
是蛋白质中原子(元素类型Ts)与配体中原子(元素类型Tt)之间的接触数总和,其中 是元素类型为Ts的原子总数, 是元素类型为Tt的原子总数。原子r和l的距离dr,l在d0+(k-2)δ和d0+(k-1)δ范围内,接触数cr,l为1,否则为0。研究中,“壳”的数量(N)为60,厚度d0=1.0×10-10 m,δ=0.5×10-10 m,最远边界(k=60)到中心原子的距离为30.5×10-10 m。一个蛋白质-配体复合物的原子接触总共产生3 840个特征。
2.2.2 LSTM与Spatial-Attention模型构建
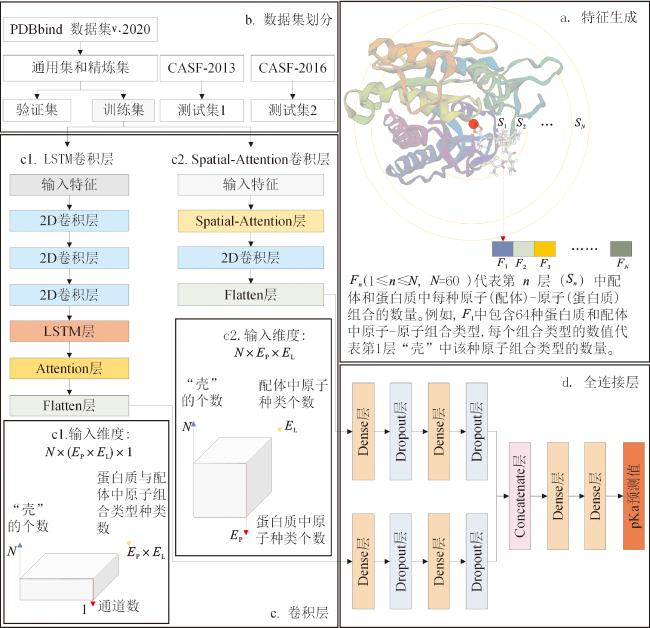
模型由卷积和全连接网络块组成,卷积网络块嵌入LSTM和Spatial-Attention模块并行。模型具体流程如图2所示。a模块从蛋白质-配体复合文件中提取蛋白质-配体原子的接触特征。b模块划分数据集,去除冗余数据。训练集提取的蛋白质-配体接触特征重塑维度后传递到c模块的c1和c2卷积网络块,c1网络块中LSTM模块能够发现输入特征中长距离的序列信息,c2网络块中Spatial-Attention模块能够发现输入特征局部信息。卷积网络块的计算结果生成向量传递到d模块,输出模型的预测值。
2.2.3 超参数设置
模型的超参数为卷积层输入维度、激活函数、学习率(lr)、衰减常数(decay)、动量(momentum)和丢失概率(dropout)。c1模块输入维度为(60,64,1),其中“壳”的层数为60,每层“壳”中原子-原子组合种类数为64,通道数为1;c2模块输入维度为(60,8,8),“壳”的层数为60,蛋白质和配体中原子种类数为8。卷积层和密集层采用ReLU激活函数。梯度下降算法可以通过迭代的方法,不断调整模型参数的取值,根据当前模型参数的取值计算出损失函数对每个参数的偏导数,按照负梯度方向更新参数,使损失函数逐渐减小,直到达到损失函数的最小值。并且梯度下降算法能够在大量数据上进行优化,并收敛到全局最优解,因此本实验整体选择随机梯度下降(SGD)优化器作为模型的最佳权重,学习率(lr)设为0.03,衰减常数(decay)设为1×10-6,动量(momentum)设为0.9。实验发现d模块中Dropout层的丢失概率(dropout rate)设置为0.1时,模型达到最高预测精度和最快收敛速度。对于每种情况下的预测值进行5次独立训练获得预测的平均值。
为了评估DLLSA模型的性能,本实验采用自定义的损失函数:
l=α(1-R)+(1-α)ERMS。
式中:R和ERMS代表Pearson(R)相关系数和均方根误差;α(0≤α≤1)是调整R和ERMS权重的参数,最终设置为0.7。
2.2.4 评估指标
结合亲和力pKa(记作Pka)表示为Kx的负对数,定义如下:
Pka=-lg Kx,
其中Kx代表抑制常数(Ki)、解离常数(Kd)或半抑制浓度(IC50)。
评价指标的定义如下:
ERMS= ;
EMA= | - |;
DS= ;
R= ;
IC= h(- );
h(x)=
式中:N代表蛋白质-配体复合物的数量; 和 是蛋白质-配体复合物的预测和真实结合亲和力值;a和b是预测和真实结合亲和力之间回归线的斜率和截距; 和 是预测值和真实结合亲和力的标准差,predicti是较大实际亲和力的预测值,predictj是较小实际亲和力的预测值。
3 结果与讨论
3.1 实验结果
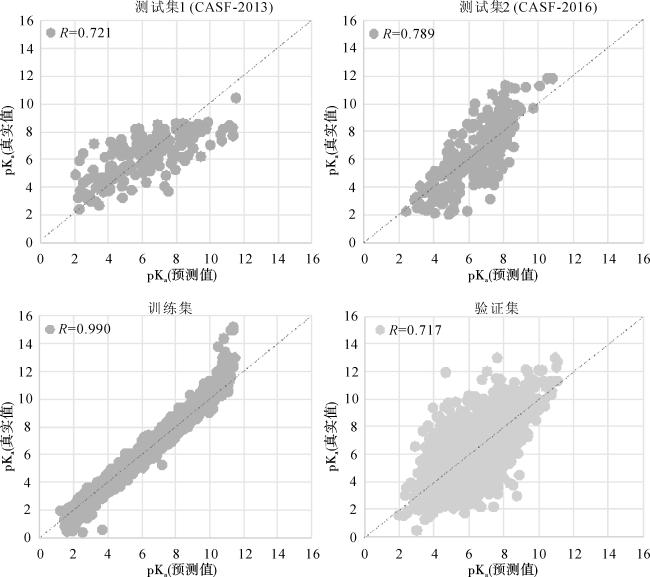
模型在不同数据集的表现如表1所示。表1中指标RMSE、MAE和SD越低,模型精度越高,R和CI指标越大模型预测结果越准确,训练集的指标均为最低,验证集和测试集的指标是模型训练获得的最低值,每个数据集的R和CI值均大于0.7。图4中每个点代表蛋白质-配体复合物的亲和力,横纵坐标分别为预测和真实的结合亲和力值。如图3所示,大多数点都分布在真实值与预测值的对角线附近,数据分布越集中在对角线,说明预测值越接近真实值,R值越高,相关性越大。图中4个数据集都呈现高于0.7的R值,表明数据集的预测和真实值之间都呈现高度相关性。CI的数值反映预测值与实际值之间的一致性,模型每个数据集的CI值都大于0.7,表明模型预测值与实际值具有高度准确性。
表1 DLLSA模型在不同数据集上的表现Tab.1 Performance of DLLSA model on different datasets |
| 数据集 | Pearson(R) | CI | RMSE | MAE | SD |
|---|---|---|---|---|---|
| 训练集 | 0.990 | 0.962 | 0.276 | 0.201 | 0.269 |
| 验证集 | 0.717 | 0.759 | 1.343 | 1.025 | 1.339 |
| CASF-2013 | 0.721 | 0.765 | 1.564 | 1.253 | 1.567 |
| CASF-2016 | 0.789 | 0.795 | 1.356 | 1.059 | 1.349 |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
表2 基准集CASF-2016在不同模型上的表现Tab.2 Performance of the benchmark set CASF-2016 on different models |
表3 基准集CASF-2013在不同模型上的表现Tab.3 Performance of the benchmark set CASF-2013 on different models |
| 数据集 | 模型 | MAE(↓) | RMSE(↓) | R(↑) | SD(↓) | CI(↑) |
|---|---|---|---|---|---|---|
| CASF-2013 | OnionNet[34] | 1.423 | 1.890 | 0.555 | 1.872 | - |
| Pafnucy[35] | 1.503 | 1.862 | 0.592 | 1.815 | - | |
| PLEC[36] | 1.246 | 1.615 | 0.716 | 1.571 | - | |
| KNN-Score[37] | - | - | 0.672 | 1.650 | - | |
| X-Score[37] | - | - | 0.614 | 1.786 | - | |
| ChemScore[37] | - | - | 0.592 | 1.822 | - | |
| ChemPLP[37] | - | - | 0.579 | 1.846 | - | |
| AutoDock Vina[38] | - | - | 0.540 | 1.901 | - | |
| AutoDock[38] | - | - | 0.541 | 1.910 | - | |
| DeepDTAF | 1.737 | 2.103 | 0.608 | 1.787 | 0.717 | |
| DLLSA | 1.253 | 1.564 | 0.721 | 1.567 | 0.765 |
3.2 消融实验
模型在CASF基准集上进行了消融实验,以评估不同模块对模型性能的影响,如表4所示。模型的输入描述符记为O,LSTM模块记为L,Spatial-Attention模块记为SA。(O+L)和(O+SA)表示实验在以描述符为输入的卷积网络上添加LSTM模块和Spatial-Attention模块,(O+L+SA)表示本文模型。表中数据显示,描述符单独使用时,性能最差。(O+L)的实验结果显著优于(O+SA)的实验结果,表明LSTM模块对描述符长序列信息的获取能够提高模型预测蛋白质-配体结合亲合力的准确性,Spatial-Attention模块在描述符上关注局部结合信息的同时限制了模型对描述符整体信息的获取。(O+L+SA)的实验结果最优,LSTM和Spatial-Attention模块的并行使得模型获取描述符的长序列非局部信息和重要的局部结合信息,两个模块达到了功能互补的效果。
表4 数据集CASF进行消融实验的结果Tab.4 Results of ablation experiments on dataset CASF |
| 数据集 | 模块组合 | MAE(↓) | RMSE(↓) | R(↑) | SD(↓) |
|---|---|---|---|---|---|
| CASF-2013 | O | 1.423 | 1.890 | 0.555 | 1.872 |
| O + L | 1.233 | 1.535 | 0.737 | 1.528 | |
| O + SA | 1.555 | 1.874 | 0.573 | 1.855 | |
| O + L + SA | 1.233 | 1.523 | 0.740 | 1.567 | |
| CASF-2016 | O | 1.137 | 1.542 | 0.707 | 1.539 |
| O + L | 1.058 | 1.358 | 0.788 | 1.353 | |
| O + SA | 1.372 | 1.694 | 0.673 | 1.624 | |
| O + L + SA | 1.056 | 1.356 | 0.789 | 1.349 |
3.3 讨论
本文提出一个新颖的深度学习模型(DLLSA),基于长短期记忆(LSTM)和注意力机制(Attention)来预测蛋白质-配体的结合亲和力,其特点在于:1)LSTM模块有效关联特征长距离序列之间的潜在关系;2)注意力机制模块改变特征局部权重,突出蛋白质-配体结合部位局部信息,抑制相对无效信息;3)深度学习技术在非线性变换揭示隐藏信息时存在黑箱决策问题,注意力机制的引入为深度学习获取关键信息提供了一定的解释性。但在本研究中,我们使用蛋白质-配体中原子的距离作为蛋白质-配体接触特征,较少关注蛋白质的序列信息与理化性质。蛋白质的表示很重要,它可以有效改进模型预测性能,也有助于揭示以前未发现的生物潜在特征,还可以作为全局特征预测蛋白质-配体远距离的间接相互作用。蛋白质的生物活性信息也值得进一步研究。因此,研究蛋白质-配体的结构和生物活性数据特征是一个重要的研究方向。在未来的研究中,我们将进一步结合蛋白质-配体的结构特征与生物活性或序列信息实现蛋白质-配体结合亲和力的预测。
4 结语
本实验采用距离“壳”中的原子接触作为指纹特征,与其他基于复杂特征的ML或CNN模型相比,具有直观简洁的特点。蛋白质-配体中原子的距离接触和化学元素类型是需要考虑的信息。类“壳”描述符能够捕捉蛋白质-配体之间非局部相互作用和局部相互作用。DLLSA在CASF基准集上进行测试,与现有经典模型进行比较,DLLSA模型优于OnionNet、Pafnucy与PLEC模型。通过消融实验验证LSTM和注意力机制嵌入深度卷积网络能有效提高预测蛋白质-配体结合亲和力的准确性,注意力机制的应用能够为深度学习黑箱决策提供有力解释。此外,蛋白质序列信息和配体SMILES序列也可以作为全局信息预测远距离间接相互作用。在今后的研究中,可以结合蛋白质-配体的结构信息与序列信息共同预测蛋白质-配体的结合亲合力。