

低级别胶质瘤(low-grade glioma,LGG)是世界卫生组织(WHO)分类为Ⅱ和Ⅲ级的、源自神经胶质细胞的颅内肿瘤,具有高度的异质性和很强的侵袭性,导致完全切除肿瘤变得困难[1-2]。LGG一般有两种分型方式:按照肿瘤恶性程度分为低、高阶两种不同的亚型;根据异柠檬酸脱氢酶(isocitrate dehydrogenase,IDH)基因突变和染色体臂1p和19q (1p/19q)的共缺失分为少突胶质瘤(IDH mutant with 1p/19q co-deletion, IDHmut-Codel)、突变型星形细胞瘤(IDH mutant without 1p/19q co-deletion, IDHmut-Non-codel)和野生型星型细胞瘤(IDH wild-type, IDH wt)三种分子亚型,不同的分子亚型具有不同的临床、病理和分子特征。研究表明,并非所有亚型都能够通过更大范围的切除来改善预后,分子亚型的不同决定患者手术切除肿瘤的范围,这直接影响患者的治疗效果[3-4]。因此,LGG分子亚型的准确分类有助于了解LGG患者的发病机制,为疾病的早期诊断提供新的思路,可以更好地指导患者的预后预测和精准靶向治疗。
随着高通量测序技术的发展,大量的多组学数据被获取[5],包括DNA甲基化、mRNA表达和miRNA表达数据等,不同类型的组学数据从不同分子层面揭示了肿瘤发生和发展的潜在机制,为人类复杂疾病的基础数据分析提供坚实基础[6]。Matsui等引入残差网络提出了一种从多模态医学图像中提取图像和数值混合数据的深度学习模型,利用该模型将LGG分为IDHmut-Codel、IDHmut-Non-codel和IDH wt三种分子亚型,获得了较好的分类性能,具有一定的临床应用价值,表明在术前对分子亚型的分类能有效帮助医生制定治疗策略[1]。然而,要确保LGG患者医学影像数据能支持准确的亚型分类耗时耗力,模型又很难在小样本数据集上学习LGG分子亚型之间的微小差异,这就会降低模型的分类性能[7-8]。因此,本文将对LGG的DNA甲基化、mRNA表达和miRNA表达数据进行联合分析,挖掘不同分子层面的关键基因,从而对LGG的分子亚型进行分类研究,进一步理解LGG的发病机制。
在LGG分子亚型的三分类中,IDH 突变和染色体臂1p/19q缺失的分子基因型均与LGG 患者的预后高度相关,与其他两种星形细胞瘤相比,少突胶质瘤对化疗放疗更敏感[4]。Ding等将图卷积网络以及视图相关性发现网络相结合,集成LGG的DNA甲基化、mRNA表达和miRNA表达数据,将LGG患者分为Ⅱ级和Ⅲ级LGG [9]。Zhong等提出了一种使用图神经网络和视图关联发现网络(view correlation discovery network,VCDN)的深度学习模型,将LGG患者分为低、高阶LGG [10]。然而,上述研究虽然均以较好的分类性能将LGG分为两种分子亚型,但可能导致一些关键特征信息的丢失,无法识别不同亚型之间对患者的预后和治疗反应产生重要影响的潜在生物学差异,从而导致医疗人员对患者应用相对粗略的治疗方案而影响其治疗效果[11]。因此,本文利用LGG的多组学数据,将LGG分为少突胶质瘤、突变型星形细胞瘤和野生型星型细胞瘤三种不同分子亚型,通过准确的LGG分子亚型分类帮助医生更好地预测患者的生存期和疾病进展风险;根据每个亚型的分子特征,医生可以获取更有价值的预后信息,从而选择更适合患者的个性化治疗方案。
深度学习可以处理不同类型的多组学数据,并自动学习这些组学数据的特征表示,已被成功用于整合多组学异构数据集。Zhao等提出了一种可扩展的多组学深度学习框架对LGG进行生存预测,将患者样本分为具有两种不同预后状态的两个亚型,在整合多组学数据和预测生存时间的性能优于其他排名靠前的方法[12]。Lin等则运用基础深度学习模型整合乳腺癌的多组学数据,对乳腺癌进行二分类以及多分类研究,与其他使用多组学数据的方法相比,该模型在二分类和多分类上具有更好的性能[13]。在LGG组学数据中,存在与预后和生存相关的关键基因,而其他基因与预后相关性较小[14]。虽然上述研究均整合了组学数据并获得了较好的分类性能,学习到不同组学数据的一些重要特征,但它们未能充分挖掘组学数据中的潜在信息,有时甚至导致关键组学特征的丢失,进而导致模型的分类性能下降。基因注意力是一种能用于一维LGG组学数据的改进的挤压和激发(squeeze-and-excitation,SE)块,通过在组学数据训练过程中自动调节基因的权重以达到提取关键基因的效果[15⇓-17]。因此,本文引入基因注意力结合深度神经网络实现低级别胶质瘤分子亚型分类,利用基因注意力来捕捉与LGG分子亚型分类相关的组学数据特征,利用添加密集层的深度神经网络中捕获到的组学数据特征以及临床数据对LGG分子亚型分类,以获得更好的分类性能。
综上,本文提出了一种基于基因注意力和密集深度神经网络的低级别胶质瘤分类方法MODAA,其框架如图1所示。本文方法MODDA将DNA甲基化、mRNA表达和miRNA表达数据进行整合,共同构建LGG的多组学特征空间;提出基因注意力网络以增强模型对关键特征的学习能力以及泛化能力,分别学习不同组学数据的重要特征;将学习到的组学数据重要特征以及临床数据特征向量进行融合,最后采用密集深度神经网络进行LGG分子亚型三分类。实验结果表明,本文方法MODDA提高了LGG分子亚型分类的性能,揭示了潜在的生物学信息,有助于深入理解LGG分子亚型的发生机制,并为精确地临床干预和个体化治疗提供重要的参考。
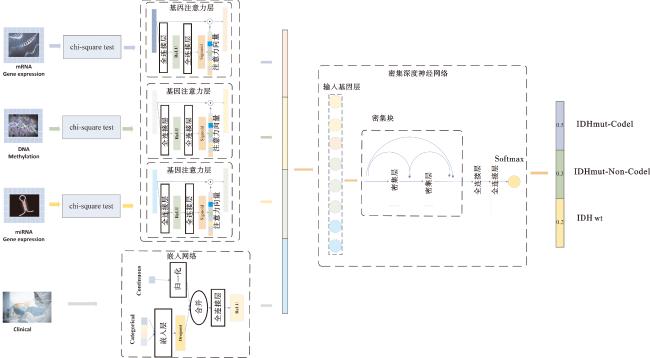
1 数据与方法
1.1 数据收集
本文使用了TCGA数据库(https://portal.gdc.cancer.gov/)提供的LGG以及用于外部验证的黑色素细胞瘤(SKCM)、卵巢癌(OV)、乳腺癌(BRCA)和肺鳞状细胞癌(LUSC)的多组学数据以及临床数据,多组学数据包括DNA甲基化、mRNA表达和miRNA表达数据。LGG、SKCM、OV、BRCA和LUSC的三种组学数据以及临床数据的详细信息如表1所示。
表1 数据集详细信息Tab.1 The details of the datasets |
| 数据集 | 特征数 | |||
|---|---|---|---|---|
| mRNA | DNA methylation | miRNA | 临床 | |
| LGG | 19 962 | 17 028 | 1 881 | 5 |
| SKCM | 22 149 | 24 533 | 827 | 5 |
| OV | 46 691 | 27 578 | 1 181 | 5 |
| BRCA | 20 531 | 22 124 | 740 | 5 |
| LUSC | 20 232 | 24 776 | 739 | 5 |
1.2 数据预处理
本文对组学和临床数据进行了预处理,预处理步骤为:1)将DNA甲基化、mRNA表达和miRNA表达数据的样本相交,获得同时包含三种组学数据的共同样本,并且获取对应样本的临床数据;2)在患者样本中,删除值为零或缺失的比例超过20%的基因;3)使用平均值填补法来解决各个组学数据中的缺失值问题。对于原始数据中的重复基因,通过计算同一基因不同表达值的平均数,并将此平均值作为该基因的最终表达值,实现了重复基因的合并。虽然LGG、SKCM、OV、BRCA和LUSC是不同类型的癌症,但它们均具有两种以上的分子亚型,表现出相似的数据分布特征,因而SKCM、OV、BRCA和LUSC数据集被用作外部验证集。为确保验证结果的可靠性,本文对所有数据集采用了统一的预处理方法。LGG、SKCM、OV、BRCA和LUSC数据集预处理后的详细信息如表2所示。
表2 预处理后的数据集详细信息Tab.2 The details of the datasets after preprocessing |
| 数据集 | 类别 | 特征数 | |||
|---|---|---|---|---|---|
| mRNA | DNA methylation | miRNA | 临床 | ||
| LGG | IDHmut-Codel:165、IDH wt:92、 IDHmut-Non-codel:239 | 16 615 | 8 049 | 434 | 5 |
| SKCM | keratin:98、immune:163、 MITF-low:58 | 20 111 | 20 055 | 528 | 5 |
| OV | MES:84、IMM:79、 DIF:98、PRO:100 | 14 850 | 13 915 | 463 | 5 |
| BRCA | Normal:115、Basal:131、 Her2:46、LumA:436、 LumB:147 | 19 227 | 20 106 | 503 | 5 |
| LUSC | Basal:32、Classical:41、Secretory:44、Primitive:33 | 20 232 | 15 510 | 739 | 5 |
1.3 卡方检验
卡方检验可以快速筛选出与目标变量相关性较高的特征,加快模型训练的速度和提高预测性能,适用于分类问题,特别是在特征数较多的情况下[20]。因此,本文使用卡方检验(chi-square test, Chi)对LGG组学数据进行特征选择,Chi用于评估两个互斥类别中的特征是否具有统计显著差异,从而筛选出与类别相关的特征。卡方检验采用公式(1)测量观察结果之间的偏差来验证所选择的特征:
C2= 。
式中Of和Ef分别表示观测频率和期望频率。
对于每种组学类型,本文分别进行卡方检验,并使用每个分类任务的相应样本,根据假设检验中的p值对特征进行排序。然后,对于每个组学数据,选择top-k特征作为深度神经网络的输入。
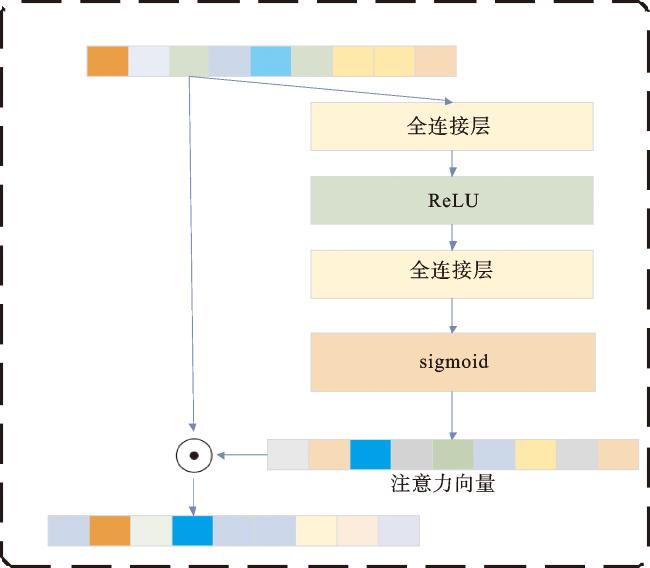
1.4 基因注意力网络
A=Sigmoid(WGA-2·ReLU(WGA-1·X0)),
X1=AT☉X0,
f(x)=max(0,x)。
式中:WGA-1∈Rq×p、WGA-2∈Rp×q是基因注意力层的权重矩阵,p是基因数量,R是真实坐标空间,q远小于p;ReLU表示修正线性单元;f(x)为其激活函数,它是一个非线性函数,可以捕获输入数据中的隐藏模式,并同时减少梯度消失;X0为基因表达量;X1是基因注意力层的输出,也是后面深度学习结构的输入。基因注意力网络中,隐藏层的维度q被设置为一个非常低的值(<100),p为组学数据中的特征数量。
如前所述,本文引入基因注意力网络,该网络使用卡方检验筛选出的基因作为输入,作为 MODDA 模型的第一层,以发现对LGG亚型分类更为关键的基因。基因注意力网络架构如图2所示。
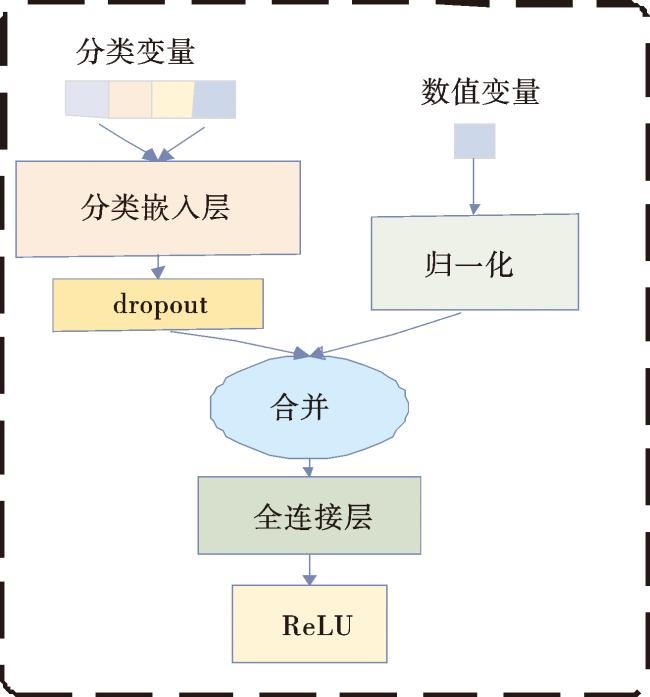
1.5 嵌入网络
不同类型的临床数据可能以不同的形式存在,包括文本、数字等,这使得数据的结构和表示方式存在差异,整合时需要克服不同数据的异构性。因此,本文使用嵌入网络来整合LGG患者临床数据中的分类变量和数值变量,对4个分类变量(即癌症类型、性别、种族和组织学类型)使用分类嵌入层和dropout将分类变量编码为数值向量;然后将经过归一化层的数值向量(即年龄)与分类变量连接起来,将连接后的向量作为全连接层(fully connected,FC)的输入,以获得固定的特征表示长度,最后经过ReLU激活函数得到嵌入网络的输出,嵌入网络的架构如图3所示。
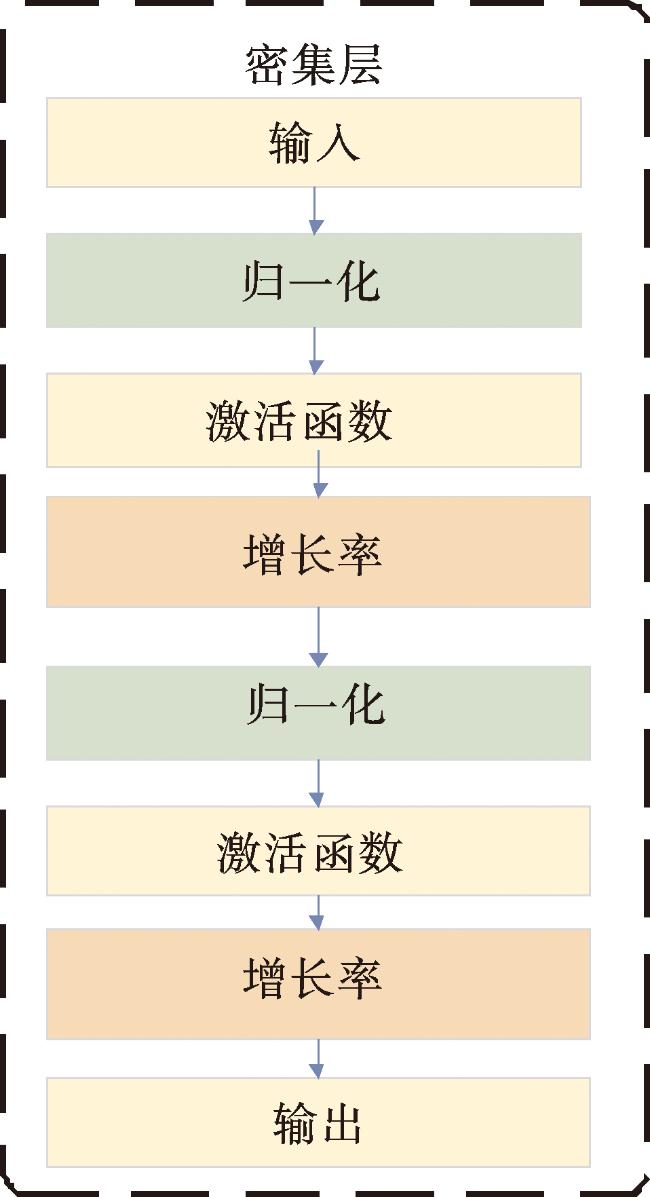
1.6 密集深度神经网络
本文提出密集深度神经网络来对LGG进行亚型分类,使用密集层作为分类模型输入层后的第一层来提取到重要的特征,密集深度神经网络由全连接层以及密集层组成。在密集层中,增长率(growth rate)定义为每个密集块内非线性变换所输出的特征数量。通过增长率的设置,可以控制每个密集块输出的特征数量,从而影响网络的深度和宽度。通常情况下,增长率越高,虽然模型的参数数量会增加,但网络的表示能力也会提高,使得模型更容易捕获复杂的特征。密集层的结构如图4所示。此外,采用包含dropout和权重衰减的分类层进行正则化,以防止过度拟合。密集层的数量基于实验结果进行选择,实验结果表明,当密集深度神经网络配置为两个密集层时,能够获得最佳的分类性能。全连接层包括1个输入层、2个隐藏层和1个输出层,对于隐藏层,第i层中的所有神经元单元都连接到上一层(i-1),然后用非线性变换函数 f 生成输出:
式中: 为隐藏层神经元数量; 是所有隐藏层神经元j的权值和偏差; 是ReLU激活函数,它是一个非线性函数,可以捕获输入数据中的隐藏模式,并同时减少梯度消失。
对于三分类,使用Softmax回归,损失函数使用交叉熵(crossentropy)损失函数。Softmax和交叉熵损失函数可以表示为
式中: 为第 个节点的输出值; 为输出节点的个数,即分类的类别个数; 表示类别的数量; 表示样本 的真实类别等于 时取1,否则取0; 表示样本 属于类别c的预测概率。
通过Softmax函数就可以将多分类的输出值转换为范围在[0,1]并且和为1的概率分布,把预测概率值最大的类别作为输出类别。
1.7 性能指标
本文验证了MODDA在LGG数据集上三分类的性能,将SKCM数据集作为外部验证数据集来验证模型三分类的泛化性能。性能评价指标包括精确度(ACC,记作ACC)、加权平均F1分数(F1_weighted) 以及宏观平均F1分数(F1_macro)。这些指标的定义如下:
ACC: 精确度代表正确预测的样本与总样本的比例。
ACC= 。
式中:TP(真阳性)表示是阳性病例并且被分类器确定为阳性病例的样本的数量;FP(假阳性)表示是阴性病例但被分类器确定为阳性病例的样本数量;FN(假阴性)表示实际上是阳性病例但被分类器确定为阴性病例的样本的数量;TN(真阴性)表示实际上是阴性病例并且被分类器确定为阴性病例的样本的数量。
F1_macro:宏观平均F1分数首先通过计算第i个类别的精确度Pi和召回率Ri,然后取平均值来计算精确度和召回率,最后得到最终的F1_macro。 、 代表第i个类别的真阳性样本数、假阳性样本数和假阴性样本数,L是总的类别数。
:加权算法是宏算法的修改版本,以解决宏算法没有考虑样本中的不平衡的问题。在计算精确度和召回率时,每个类别的精确度和召回率乘以该类别在总样本中的百分比
1.8 模型训练
本文方法MODDA是基于Torch 1.9.1实现的。在训练模型过程中,为了防止模型过拟合,本文采取了dropout、权重衰减(L2正则化)来保证模型的有效性,且将dropout率和权重衰减率分别设置为0.2和0.001。为了保证模型训练过程中的稳定性,本文将学习率、epoch、batch size分别设置为0.01、100、256。
2 实验结果与讨论
2.1 模型参数选择
2.1.1 密集层(dense layer)层数的选择
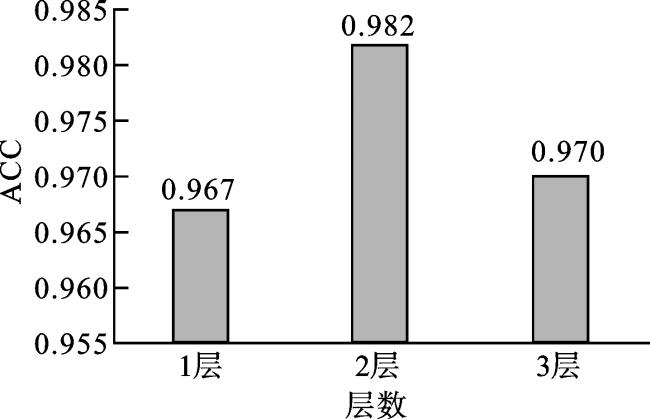
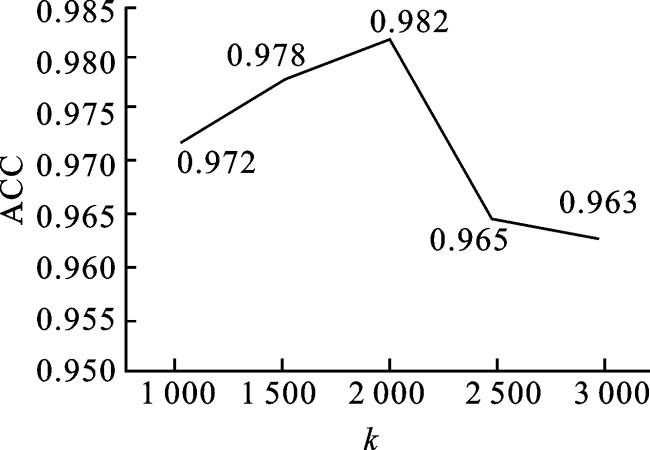
2.1.2 卡方检验中k的选择
在本文中,卡方检验被用于筛选LGG的DNA甲基化、mRNA表达和miRNA表达数据中的关键基因。因此,卡方检验中k值的选取成为选择最优特征子集的关键步骤,需要探索合适的k值,以选择到最优特征子集。在保持其他参数不变的基础上,鉴于miRNA特征的数量较少,miRNA表达数据的k值被设定为其原始特征数量,即434。对mRNA表达和DNA甲基化数据的k的取值设置为1 000、1 500、2 000、2 500和3 000,并利用本文方法MODDA 在LGG三种分子亚型的多分类任务上进行实验,选择ACC最高时的k值。结果如图6所示。在图6中,k取2 000时,ACC值为0.982,达到最优。可以看出,当k分别取1 000和1 500时,ACC呈现上升趋势,这是由于当k较小时,基因的分组较少,有一些重要基因没有被选到,不利于模型的学习。当k取2 500和3 000,选到的与LGG分子亚型相关的基因增多,但是结果没有表现得更好。这是由于当k过大时,筛选到的基因中出现冗余现象,并且数据的维度增高,从而影响到模型的性能。
2.2 单组学和多组学数据三分类的性能比较
为了验证本文方法MODDA是否能够学习到LGG组学数据的关键基因从而提高LGG分子亚型的分类性能,本文在三分类任务上设计了单组学与多组学对比实验。在实验中我们并没有在模型训练之前合并所有组学数据特征表示,而是对每种组学数据分别先使用基因注意力网络提取特征,再对提取到的特征进行拼接,最后嵌入临床数据进行分子亚型三分类,采用十折交叉验证的ACC、F1_weighted和F1_macro作为测量标准,其中ACC是精确度;F1_macro是对每类F1分数计算算术平均值,能平等地对待所有类来反映模型性能,但不考虑不同类别的重要性;F1_weighted把每个类别的样本数量作为权重,计算加权F1分数,考虑了不同类别的重要性来评估三分类性能[27]。LGG单组学和多组学数据三分类的对比实验结果如图7所示。
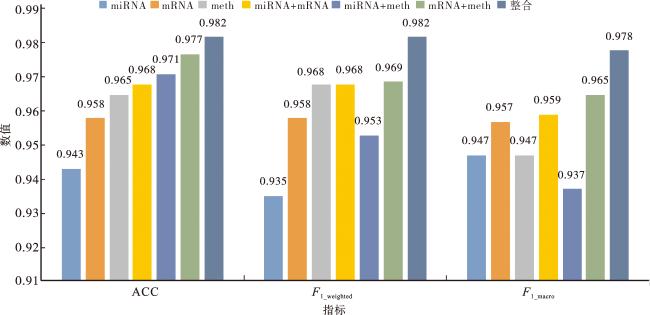
从图7中可以看到,与使用单组学数据相比,将DNA甲基化、mRNA表达和miRNA表达数据进行整合,模型的整体分类性能均有较为明显的提升,分类的ACC、F1_weighted和F1_macro的值分别为0.982、0.982和0.978,均比其他情况的分类性能要好。与单一组学miRNA表达、mRNA表达以及DNA甲基化数据相比,本文利用多组学在ACC上分别增加了3.9%、2.4%、1.7%。与两种组学组合的情况对比,本文采用的三组学组合方式的ACC高了1.4%、1.1%、0.5%。这些结果表明,本文方法MODDA能够很好地学习到每个组学数据的高级特征表示,能够有效避免不同组学数据之间的相互干扰且提取到组学数据的关键基因。并且DNA甲基化、mRNA表达和miRNA表达数据有密切的关联,整合三种组学数据能够补充提取单组学数据特征时丢失的信息,使得模型对LGG分子亚型的识别率得到较大的提升[28]。可以观察到,去除DNA甲基化数据时ACC的下降幅度最大,从0.982下降为0.968,而去除miRNA表达数据和mRNA的ACC的下降幅度则较小,分别下降了0.5%和1.1%。首先是DNA甲基化数据,其次是mRNA表达数据,可以更好地区分LGG的分子亚型,而miRNA表达数据对LGG分子分型的贡献最小,这是由于在LGG细胞中DNA甲基化与LGG的分子亚型有较强的关联,而miRNA和mRNA只提供了小部分的关于LGG分子亚型的信息[29]。也正是如此,将miRNA表达、mRNA表达以及DNA甲基化数据进行整合提高了模型对LGG分子亚型的分类能力,ACC为0.982。
2.3 消融实验
为了验证LGG组学数据层面的合并与特征层面的合并对分类性能的影响以及MODDA中不同模块对分类性能提升的作用,本文进行了消融实验。DNN能够自动从原始数据中学习特征,而无需手动设计或选择特征,这使得模型能够更好地适应不同类型的数据,尤其是对于复杂的、高维度的数据。因此,本文将DNN作为基准分类模型,验证在此基础上融合不同的模块和临床数据对分类性能的影响,以及使用不同组学数据组合的性能比较。采用十折交叉验证的ACC、F1_weighted和F1_macro作为测量标准。本文使用基因注意力网络提取每种组学的重要特征,嵌入网络将临床数据分类变量和连续变量融合,再与组学数据重要特征整合,整合后的特征输入密集深度神经网络进行亚型分类。DNN结合基因注意力和添加密集块在LGG不同组学数据组合情况下的亚型分类性能比较见表3。
表3 融合不同模块后不同组学数据组合的亚型分类结果Tab.3 Subtype classification results of different omics data combinations after integrating different modules |
| 设定 | 指标 | miRNA | mRNA | Meth | miRNA+ mRNA | miRNA+ Meth | mRNA+ Meth | miRNA+ mRNA+ Meth |
|---|---|---|---|---|---|---|---|---|
| DNN(no_Chi) | ACC | 0.854 | 0.876 | 0.892 | 0.899 | 0.895 | 0.905 | 0.914 |
| F1_weighted | 0.853 | 0.876 | 0.890 | 0.899 | 0.894 | 0.905 | 0.913 | |
| F1_macro | 0.847 | 0.857 | 0.884 | 0.887 | 0.873 | 0.902 | 0.908 | |
| DNN | ACC | 0.854 | 0.898 | 0.902 | 0.905 | 0.908 | 0.913 | 0.924 |
| F1_weighted | 0.853 | 0.898 | 0.9 | 0.905 | 0.906 | 0.913 | 0.924 | |
| F1_macro | 0.847 | 0.897 | 0.902 | 0.902 | 0.908 | 0.912 | 0.919 | |
| DNN+基因注意力 | ACC | 0.923 | 0.938 | 0.945 | 0.947 | 0.95 | 0.955 | 0.966 |
| F1_weighted | 0.915 | 0.938 | 0.948 | 0.947 | 0.932 | 0.947 | 0.963 | |
| F1_macro | 0.927 | 0.937 | 0.927 | 0.938 | 0.916 | 0.943 | 0.969 | |
| DNN+密集块 | ACC | 0.893 | 0.908 | 0.915 | 0.917 | 0.92 | 0.925 | 0.936 |
| F1_weighted | 0.885 | 0.908 | 0.918 | 0.917 | 0.902 | 0.917 | 0.933 | |
| F1_macro | 0.897 | 0.907 | 0.897 | 0.908 | 0.886 | 0.913 | 0.941 | |
| DNN+临床数据 | ACC | 0.903 | 0.918 | 0.925 | 0.927 | 0.93 | 0.935 | 0.946 |
| F1_weighted | 0.895 | 0.918 | 0.928 | 0.927 | 0.912 | 0.927 | 0.943 | |
| F1_macro | 0.907 | 0.917 | 0.907 | 0.918 | 0.896 | 0.923 | 0.949 | |
| DNN+基因注意力+ | ACC | 0.94 | 0.951 | 0.961 | 0.963 | 0.966 | 0.97 | 0.975 |
| 密集块 | F1_weighted | 0.94 | 0.949 | 0.958 | 0.963 | 0.965 | 0.967 | 0.975 |
| F1_macro | 0.938 | 0.957 | 0.963 | 0.958 | 0.961 | 0.963 | 0.961 | |
| DNN+基因注意力+ | ACC | 0.933 | 0.948 | 0.955 | 0.957 | 0.96 | 0.965 | 0.972 |
| 密集块 | F1_weighted | 0.925 | 0.948 | 0.958 | 0.957 | 0.942 | 0.957 | 0.971 |
| F1_macro | 0.937 | 0.947 | 0.937 | 0.948 | 0.926 | 0.953 | 0.968 | |
| DNN+密集块+ | ACC | 0.903 | 0.92 | 0.918 | 0.925 | 0.927 | 0.93 | 0.931 |
| 临床数据 | F1_weighted | 0.895 | 0.92 | 0.918 | 0.928 | 0.909 | 0.922 | 0.928 |
| F1_macro | 0.907 | 0.911 | 0.917 | 0.907 | 0.893 | 0.918 | 0.919 | |
| DNN+基因注意力+ | ACC | 0.943 | 0.958 | 0.965 | 0.968 | 0.971 | 0.977 | 0.982 |
| 密集块+临床数据 | F1_weighted | 0.935 | 0.958 | 0.968 | 0.968 | 0.953 | 0.969 | 0.982 |
| F1_macro | 0.947 | 0.957 | 0.947 | 0.959 | 0.937 | 0.965 | 0.978 |
表3中不同的行表示采用不同的单一组学数据或多种组学数据组合。DNN(no_Chi)表示在模型训练之前将特征表示直接合并,不对所有组学数据进行特征选择,进行LGG分子亚型分类。从表3中可以看出,相对于特征层面合并的消融实验,DNN(no_Chi)获得了较差的分类性能,DNN分别融合基因注意力以及添加密集块之后均能提升分类性能,使用基因注意力分别结合嵌入模块以及密集块的分类性能较好且都有提升,并且单引入基因注意力模块要比引入密集块或者临床数据的性能更好,ACC、F1_weighted和F1_macro值为0.966、0.963和0.969,并且比单引入密集层要分别高出3%、3%和2.8%。首先这是因为利用基因注意力网络学习每个组学数据的高级特征表示能够有效提取到与分类相关的组学数据特征,从而提升分类的性能;密集块用于增强特征重用和梯度流动,从而提高模型的性能和泛化能力,本模型中用于防止在分类网络中特征信息向下一层传递丢失太多信息,关注特征的重用和信息的共享,因而能帮助提升模型精度[30]。并且同时融合这两个模块能获得更好的分类性能。临床数据使用嵌入模块与多组学数据进行整合,也就是在特征整合过程中补充LGG的临床信息,而临床信息能提供分子亚型的更多信息,从而提升分类性能[31],因而融合这些模块的基础上再引入临床数据能够获得更好的分类性能,ACC、F1_weighted和F1_macro值为0.982、0.982和0.978。
表3中不同的列表示采用不同的单一组学数据或多种组学数据组合,2~4列表示采用单一组学数据,5~7列表示整合两两组合的组学数据,最后一列表示采用三种组学数据。记录了融合不同模块的情况下整合不同组学组合数据取得的ACC、F1_weighted和F1_macro值。从表3看出,与单一组学的情况或两种组学组合的情况相比,本文采用三组学组合方式取得了最高的ACC、F1_weighted和F1_macro值,并且在融合了所有提出模块的情况下,整合多组学数据进行LGG分子亚型分类获得了最好的性能,ACC、F1_weighted和F1_macro值分别为0.982、0.982和0.978。ACC的值比使用单组学miRNA、mRNA和DNA甲基化数据分别提高了3.9%、2.4%和1.7%;ACC的值分别比组学两两组合提升了1.4%、1.1%和0.5%。这也验证了融合上述模块整合多组学的有效性,模型具有较好的分类性能。
2.4 对比实验
为了评估本文方法MODDA的分类性能,本文基于LGG数据集将MODDA与其他现有方法的分类性能进行了比较。在这部分实验中,我们选择了7种具有代表性的方法与MODDA进行了比较。其中,KNN和SVM是传统机器学习算法,它们在许多分类任务中都表现出了良好的性能,并且目前没有研究者直接使用机器学习方法整合LGG多组学数据进行LGG分子亚型分类。DeepMO、P-NET、MOMA、MOGONET和MODILM是5种深度学习算法,DeepMO利用编码子网络融合多组学数据,在乳腺癌亚型多分类任务中获得了较好的分类性能,作为一个基础深度学习对比方法。MOMA、MOGONET和MODILM是当前在整合多组学数据进行LGG二分类任务中表现最佳的深度学习方法。P-NET是一个基于生物学原理的深度学习模型,它有效地将生物学中的分层知识融合入神经网络架构中,减少了模型学习的参数数量,并已在前列腺癌(PARD)、炎症性肠病(IBD)和结直肠癌(CRC)等数据集上展现出优秀的分类性能。因此,选取以上方法用于LGG分子亚型三分类对比实验。表4给出了本文方法MODAA与选取的对比方法在LGG数据集上的性能比较。
表4 不同分类方法在LGG数据集分类性能结果Tab.4 classification performance results of different classification methods in the LGG dataset |
| 方法 | ACC | F1_weighted | F1_macro |
|---|---|---|---|
| SVM | 0.767 | 0.770 | 0.742 |
| KNN | 0.751 | 0.751 | 0.766 |
| DeepMO | 0.821 | 0.821 | 0.813 |
| P-NET | 0.922 | 0.922 | 0.913 |
| MOMA | 0.939 | 0.939 | 0.923 |
| MOGONET | 0.943 | 0.947 | 0.928 |
| MODILM | 0.975 | 0.960 | 0.945 |
| MODAA | 0.982 | 0.982 | 0.978 |
实验结果表明,本文方法MODDA在LGG数据集上的ACC、F1_weighted和F1_macro值分别为0.982、0.982和0.978,取得了较好性能。具体而言,在对比方法中,SVM在机器学习方法中表现最好,ACC、F1_weighted和F1_macro三个指标分别为0.767、0.770和0.742,DeepMO在深度学习方法中表现最差,三项指标分别为0.821、0.821和0.813,但DeepMO的ACC、F1_weighted和F1_macro比SVM分别高出6%、6%和7.1%,亦即在对比方法中深度学习方法比机器学习方法的性能要更好,这是因为深度学习模型通常包含大量参数和非线性激活函数,使其能够建模高度非线性的数据关系,处理复杂的数据模式和结构时具有优势。相对于MOMA、MODILM和MOGONET这4种深度学习方法,本文方法MODDA在ACC、F1_weighted和F1_macro三个指标上分别优于MOMA 4.3%、4.3%和5.5%,优于MOGONET 3.9%、3.5%和5%,优于MODILM 0.7%、2.2%和3.3%,优于P-NET 6%、6%和6.5%。我们可以观察到MODDA在分类方面优于其他方法。这是因为本文提出的方法MODDA综合了多个模块,从而能够提升模型的整体分类性能,基因注意力模块在能够关注到与LGG分子亚型分类相关的特征的同时,利用密集块减少分类网络中的信息丢失,而嵌入模块补充了LGG分子亚型的重要临床数据信息,从而提高了模型的分类性能。本文是进行LGG分子亚型三分类研究,相对于分子亚型二分类,需要处理更复杂的组学数据分布以及预后信息,并且取得了较好的分类性能。
2.5 外部数据集验证MODDA分类的泛化性能
为了研究本文方法MODDA的泛化性能,在SKCM、OV、BRCA和LUSC数据集上进行了单组学与多组学对比实验。针对每种组学数据,本文方法MODDA首先使用基因注意力网络提取特征,再对提取到的特征进行拼接,最后嵌入临床数据进行分子亚型三分类,采用十折交叉验证的ACC、F1_weighted和F1_macro作为评价标准,不同类型的组学数据对SKCM、OV、BRCA和LUSC亚型分类的ACC、F1_weighted和F1_macro见表5。从表5中可以看出,相对于单组学多分类,在SKCM、OV、BRCA和LUSC多组学数据的ACC、F1_weighted和F1_macro都有提升,这验证了本文方法MODDA整合多组学数据的有效性。
表5 不同癌症数据集的分类结果Tab.5 Classification results of different cancer datasets |
| 数据集 | 指标 | miRNA | mRNA | Meth | miRNA+ mRNA | miRNA+ Meth | mRNA+ Meth | miRNA+ mRNA+ Meth |
|---|---|---|---|---|---|---|---|---|
| SKCM | ACC | 0.889 | 0.905 | 0.913 | 0.919 | 0.928 | 0.939 | 0.942 |
| F1_weighted | 0.897 | 0.891 | 0.913 | 0.920 | 0.927 | 0.932 | 0.939 | |
| F1_macro | 0.879 | 0.886 | 0.912 | 0.902 | 0.925 | 0.926 | 0.928 | |
| OV | ACC | 0.864 | 0.878 | 0.892 | 0.903 | 0.917 | 0.923 | 0.933 |
| F1_weighted | 0.863 | 0.878 | 0.892 | 0.902 | 0.917 | 0.923 | 0.930 | |
| F1_macro | 0.857 | 0.877 | 0.890 | 0.902 | 0.910 | 0.912 | 0.925 | |
| BRCA | ACC | 0.823 | 0.838 | 0.845 | 0.847 | 0.850 | 0.855 | 0.884 |
| F1_weighted | 0.823 | 0.838 | 0.845 | 0.847 | 0.852 | 0.855 | 0.879 | |
| F1_macro | 0.817 | 0.837 | 0.827 | 0.838 | 0.846 | 0.848 | 0.843 | |
| LUSC | ACC | 0.873 | 0.888 | 0.895 | 0.897 | 0.900 | 0.907 | 0.915 |
| F1_weighted | 0.865 | 0.888 | 0.898 | 0.897 | 0.900 | 0.907 | 0.915 | |
| F1_macro | 0.867 | 0.887 | 0.887 | 0.888 | 0.896 | 0.903 | 0.913 |
在SKCM、OV、BRCA和LUSC数据集上,将本文方法MODDA与其他方法进行了进一步的对比实验,结果如表6所示。结果表明,本文方法MODDA在上述4个数据集上的ACC、F1_weighted和F1_macro指标都优于对比方法。在现有的方法中,MODILM在所有数据集上的分类性能都是最好的,而与MODILM相比,本文方法MODDA在多分类任务上的ACC、F1_weighted和F1_macro方面分别获得了0.3%到15.5%、0.2%到17.7%和0.1%到18.3%的改进。实验结果也表明,本文方法MODDA能较好地整合多组学数据,并且取得了较好的分类性能,进一步验证了本文方法对癌症分子亚型分类的有效性和泛化性。
表6 不同方法在不同癌症数据集上的分类结果比较Tab.6 Comparison of classification results of different methods on different cancer datasets |
| 方法 | SKCM | OV | BRCA | LUSC | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ACC | F1_weighted | F1_macro | ACC | F1_weighted | F1_macro | ACC | F1_weighted | F1_macro | ACC | F1_weighted | F1_macro | |
| SVM | 0.813 | 0.812 | 0.805 | 0.831 | 0.831 | 0.827 | 0.729 | 0.702 | 0.64 | 0.774 | 0.771 | 0.754 |
| KNN | 0.852 | 0.852 | 0.835 | 0.853 | 0.853 | 0.853 | 0.782 | 0.782 | 0.78 | 0.776 | 0.776 | 0.758 |
| DeepMO | 0.879 | 0.873 | 0.859 | 0.897 | 0.895 | 0.887 | 0.789 | 0.787 | 0.775 | 0.785 | 0.785 | 0.782 |
| P-NET | 0.905 | 0.903 | 0.901 | 0.901 | 0.901 | 0.899 | 0.816 | 0.814 | 0.809 | 0.84 | 0.838 | 0.834 |
| MOMA | 0.908 | 0.905 | 0.908 | 0.912 | 0.911 | 0.908 | 0.829 | 0.829 | 0.824 | 0.851 | 0.851 | 0.847 |
| MOGONET | 0.919 | 0.919 | 0.917 | 0.92 | 0.919 | 0.917 | 0.836 | 0.832 | 0.815 | 0.853 | 0.853 | 0.842 |
| MODILM | 0.928 | 0.927 | 0.927 | 0.93 | 0.93 | 0.928 | 0.845 | 0.84 | 0.804 | 0.865 | 0.855 | 0.833 |
| MODAA | 0.942 | 0.939 | 0.928 | 0.933 | 0.93 | 0.925 | 0.884 | 0.879 | 0.843 | 0.915 | 0.915 | 0.913 |
2.6 基因富集分析
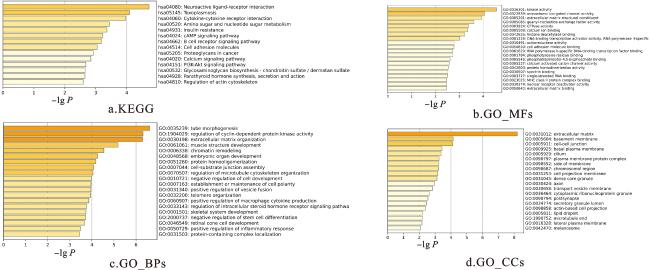
为了进一步了解LGG样本之间的差异,本文对选择出来的对LGG分子分型贡献较大的组学数据,即DNA甲基化和mRNA基因表达数据,利用卡方检验选择的前2 000个基因进行富集分析。本文使用了人类基因的整个集合作为背景,使用Metascape进行富集分析,涵盖了基因本体(Gene Ontology,GO)和京都基因与基因组百科全书(Kyoto Encyclopedia of Genes and Genomes,KEGG)通路数据库,以了解LGG分子亚型之间的差异。KEGG通路富集结果如图8a所示;GO的三个富集通路分子功能(表示为GO_MFs)、生物过程(表示为GO_BPs)和细胞组分(表示为GO_CCs)富集结果如图8b~8d所示。
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
实验结果表明,在KEGG富集结果中显著富集的通路包括神经活性配体-受体相互作用和细胞因子-细胞因子受体相互作用。GO富集中最显著富集的分子功能(GO_MFs)、生物过程(GO_BPs)和细胞组分(GO_CCs)分别是激酶活性、管状形态发生以及细胞外基质,并且生物过程细胞周期蛋白依赖性激酶活性的调控也较为显著。LGG的生存率较低有一个预后分子标记,称为SYT16表达。LGG中受SYT16调节的关键途径包括神经活性配体-受体相互作用、钙信号传导途径和T细胞增殖[32]。研究表明,GMFG在LGG中显著上调,这预示着LGG的无病生存期较差,并且GMFG的潜在的作用机制之一就是细胞因子-细胞因子受体相互作用[33]。LGG患者治疗后所产生的激酶活性差异较大,并且细胞周期蛋白依赖性激酶抑制2C(CDKN2C)表达升高与LGG患者预后较差相关,利用这一特点可以评估LGG患者治疗的初步疗效[34-35]。Sun等通过研究得出管状形态发生是LGG和肺癌发生的关键因素之一[36-37]。LGG中的特异性与细胞外基质(ECM)有关,这表现为较差的生存率。因此,这些生物通路都在低级别胶质瘤的发生过程中发挥着调控作用。
3 结语
本文提出了具有基因注意力的多组学数据整合方法MODDA,用于LGG分子亚型分类。该方法使用基因注意力网络提取重要特征,嵌入网络将临床数据的分类变量和数据变量编码融合得到临床数据特征,之后与提取到的组学数据特征进行融合,采用密集深度神经网络进行三分类。结果表明,整合多组学数据可以提高模型对LGG分子亚型分类的性能,通过对SKCM、OV、BRCA和LUSC数据集的外部验证表明了该模型具有泛化性。此外,本文方法MODDA三分类性能方面也优于其他现有方法。同时,通过对重要基因及其通路进行分析,对LGG分子亚型之间差异进行了生物学解释。尽管本文方法MODDA在LGG分子亚型分类中表现出较好的性能,但仍需要进一步改进。在未来的工作中,除了使用的三种类型的组学数据之外,MODDA 还可以扩展以集成日益复杂的数据,例如蛋白质组数据,为探索LGG分子亚型的生物学模型提供有力指导。