

随着化石能源的不断消耗,环境问题和能源危机日益严重,寻找可持续的清洁能源来代替化石能源是当务之急。氢气作为一种清洁高效的能源,具有利用率高、热值高和来源丰富等特点,在环境要求和能源需求方面展现出巨大的潜力,是最有可能代替传统化石燃料的能源之一[1⇓-3]。电催化制氢是利用电能来分解水制取氢气,这种方法简单可靠,所需电力可通过太阳能、风能和核能等可再生能源来获得,而且水资源丰富,所以电解水是有望实现工业化生产氢气的方法之一[4-5]。但是,电解水制氢的效率受制于催化剂的催化性能。所以,高效稳定的廉价析氢催化剂的开发对电解水制氢产业尤为重要[6]。
二维材料,如石墨烯、氮化硼、过渡金属硫族化合物等,因具有较大的比表面积、特定的暴露晶面、特殊的尺寸效应等特点,从而表现出良好的催化活性、良好的产物选择性以及优异的稳定性,被认为是一类极具潜力的析氢催化剂[7⇓-9]。其中,以MoS2为代表的过渡金属硫化物具有类石墨烯结构和优异的物理化学性质,被广泛地应用在电催化析氢研究中[10⇓⇓⇓⇓-15]。单层无缺陷的本征MoS2具有化学惰性,需要通过改性手段进行调控。掺杂是一种有效的改性手段,掺杂元素与载体之间的相互作用可以通过原子轨道的杂化和电荷转移来调整载体的催化活性。因此,向MoS2掺杂其他元素,可以有效调整其电催化析氢活性[16⇓⇓⇓-20]。
然而,对MoS2掺杂的研究[21-22]仍存在很多不足。例如:未系统地研究掺杂元素、掺杂方式及掺杂位点对MoS2结构及催化活性的影响。为了深入研究d带电子掺杂对MoS2的氢吸附影响规律,并以此来设计和开发新的MoS2基催化剂,本文基于密度泛函理论的第一性原理计算,系统研究了3d过渡金属(Sc、Ti、V、Cr、Mn、Fe、Co、Ni、Cu、Zn)掺杂MoS2(TM-MoS2)对结构稳定性及析氢性能的影响,分析了掺杂元素在MoS2不同位置的形成能,比较了氢在TM-MoS2表面不同活性位点的吸附能和吸附自由能,并对TM-MoS2的催化活性进行筛选,这也为MoS2基催化剂在析氢反应方面的应用提供理论参考。
1 计算方法
本文计算采用了基于密度泛函理论的VASP软件[23]。采用广义梯度近似(generalized-gradient approximation,GGA)中的Perdew-Burke-Ernzerhof(PBE)函数描述电子之间的交换相互作用[24],并采用Grimme的色散校正的密度泛函理论(density functional theory dispersion,DFT-D)进行范德华相互作用校正[25]。离子实用投影缀加波描述。所有优化都在平面波基组中进行,布里渊区K点网格设置为3×3×1,平面波截断能为400 eV。所有计算都使用自旋不受限制的方法。为了获得收敛的几何结构,原子间相互作用力收敛精度设置为0.1 eV/nm。为了避免因周期性计算方法而引入的相互作用,将z方向真空层设定为1.5 nm。
单层2H相MoS2(001)结构模型如图1所示,是由Mo原子夹在两层S原子之间组成的三明治结构,Mo和S原子排列方式类似于石墨烯的平面六角阵列。对本征MoS2(001)进行几何优化,之后将过渡金属原子吸附在MoS2表面;另一种掺杂模型是不同3d过渡金属原子取代Mo原子进行替位式掺杂。再将掺杂后的体系进行几何优化,在优化的基础上对其氢吸附自由能进行分析。
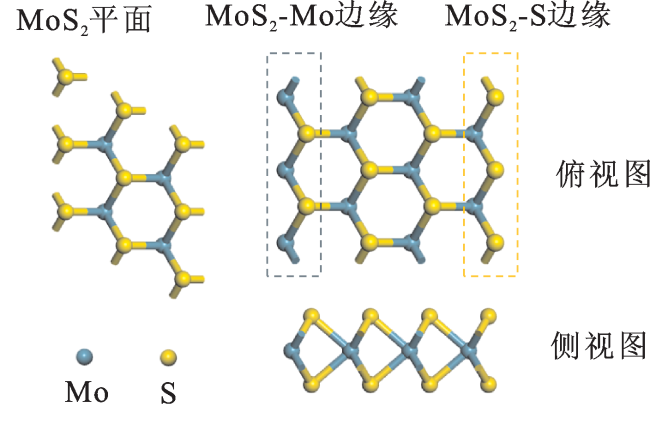
为了确定最稳定的掺杂表面结构,根据以下公式计算形成能(Ef):
Ef= - -NTMUTM+NMoUMo。
其中: 为过渡金属掺杂MoS2优化后的总能量; 是未掺杂MoS2的能量;UTM和UMo分别是过渡金属和Mo的化学势;NTM是掺入的过渡金属原子个数;NMo是被替换的Mo原子个数。金属的化学势通过计算单质的能量除以原子数来得到[26]。Ef代表掺杂的难易程度,若Ef数值为负,说明掺杂易于实现。
为了确定最稳定过渡金属吸附和氢吸附构型,根据以下公式计算吸附能(Eads):
Eads/TM= -(+ETM),
Eads/H= -(+EH)。
其中: 和 为吸附物过渡金属(TM)或氢(H)与表面结合的总能量; 和 分别为清洁表面MoS2的能量和一个过渡金属原子的能量;ETM和EH分别为清洁表面TM-MoS2的能量和气体分子H2能量的1/2;Eads/H反映了H在材料表面吸附的难易程度。根据这个定义,Eads为负值则意味着吸附为自发进行的放热反应。
为了比较不同掺杂结构MoS2的析氢活性,吸附氢吉布斯自由能变化(ΔG)按下式计算:
ΔG=ΔEads(H)+ΔEZPE-TΔS。
2 结果及讨论
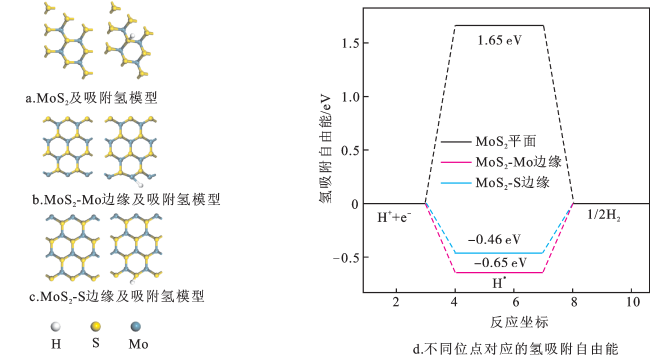
图3a为不同过渡金属(Sc、Ti、V、Cr、Mn、Fe、Co、Ni、Cu和Zn)掺杂MoS2平面的形成能。所有掺杂元素取代Mo位点在MoS2表面的Ef都是正值,说明这些掺杂MoS2平面的样品制备过程较困难。图3b为不同过渡金属掺杂MoS2平面的氢吸附自由能。过渡金属元素掺杂MoS2平面的 都小于MoS2平面的Δ (1.65 eV),说明过渡金属元素掺杂MoS2平面提高了MoS2平面的析氢活性。其中Co掺杂MoS2平面的Δ (0.01 eV)更靠近0 eV,说明Co掺杂MoS2平面具有最佳的析氢活性;然而Co吸附在MoS2表面的Ef为4.51 eV,说明Co掺杂MoS2平面的制备比较困难。图3c为不同过渡金属掺杂MoS2平面的吸附氢模型,可以看出不同过渡金属元素掺杂MoS2平面后,其结构未发生明显变化,但吸附氢的模型存在差异,说明不同过渡金属掺杂MoS2平面对其析氢活性有影响。
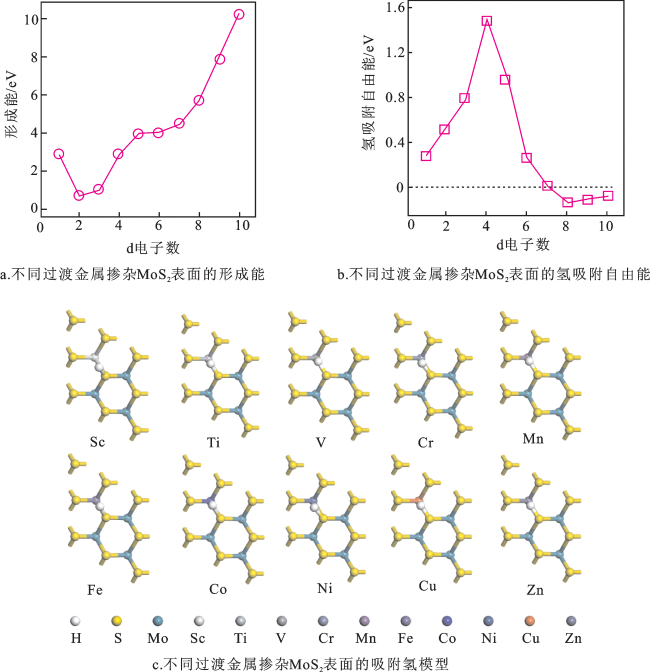
相比于本征MoS2平面,不同过渡金属掺杂MoS2平面的结构均提高了析氢性能。但由于不同过渡金属掺杂MoS2平面的Ef均为正值,说明不同过渡金属掺杂MoS2平面的制备不易实现。所以,表面掺杂对提高MoS2析氢性能并非最佳选择。
图4a为不同过渡金属吸附在MoS2表面的吸附能图。过渡金属元素吸附在MoS2表面的吸附能都为负值,是热力学自发过程。相比于其他几种元素,Ti和Ni掺杂以后,MoS2平面对氢的吸附更强(Eads/Ti=-3.06 eV,Eads/Ni=-3.06 eV)。图4b为不同过渡金属吸附在MoS2表面的氢吸附自由能。过渡金属元素吸附在MoS2表面的 都小于MoS2平面的Δ (1.65 eV),说明过渡金属元素吸附在MoS2表面提高了MoS2表面的析氢活性;其中,V吸附在MoS2表面的自由能(Δ =-0.02 eV)更加接近0 eV,说明V吸附在MoS2表面具有更高的析氢活性。结合图4a和4b,V吸附在MoS2表面可能具有最佳的理论析氢活性,尽管Ti和Ni掺杂MoS2平面的Eads更低,但Ti和Ni掺杂MoS2平面的氢吸附自由能距0 eV更远( =-3.06 eV, =-3.04 eV),说明析氢活性较差。所以,V吸附在MoS2表面有利于提高析氢反应活性。图4c为不同过渡金属吸附在MoS2表面的吸附氢模型。不同过渡金属元素吸附在MoS2平面未对其结构产生影响,而且不同元素吸附在MoS2表面的吸附氢模型也基本一致。
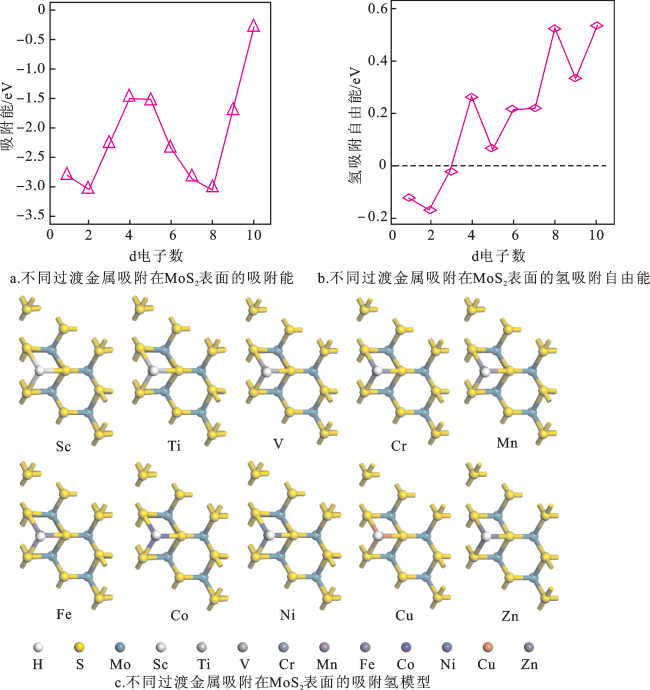
不同过渡金属吸附在MoS2表面之后,金属位点作为H吸附中心。不同金属位点对于H的吸附活性相比于本征MoS2表面有所提升,V吸附在MoS2表面具有最佳的析氢活性。
图5a为不同过渡金属掺杂MoS2的Mo边缘的形成能。Sc、Ti和V掺杂MoS2的Mo边缘的Ef为正值(Ef/Sc=0.45 eV,Ef/Ti=1.26 eV,Ef/V=0.03 eV),其他元素掺杂MoS2的Mo边缘的Ef都为负值,说明除Sc、Ti和V以外的过渡金属元素掺杂MoS2的Mo边缘制备较容易。图5b为不同过渡金属掺杂MoS2的Mo边缘之后,过渡金属位点作为H吸附中心的氢吸附自由能。Co掺杂MoS2的Mo边缘的 (0.07 eV)距离0 eV更近,说明这种掺杂结构的析氢活性更好。图5c为不同过渡金属掺杂MoS2的Mo边缘的吸附氢模型。不同过渡金属掺杂对MoS2的Mo边缘结构没有产生大的影响,但对氢的吸附有所改变,说明不同过渡金属掺杂对MoS2的Mo边缘结构的析氢活性有影响。
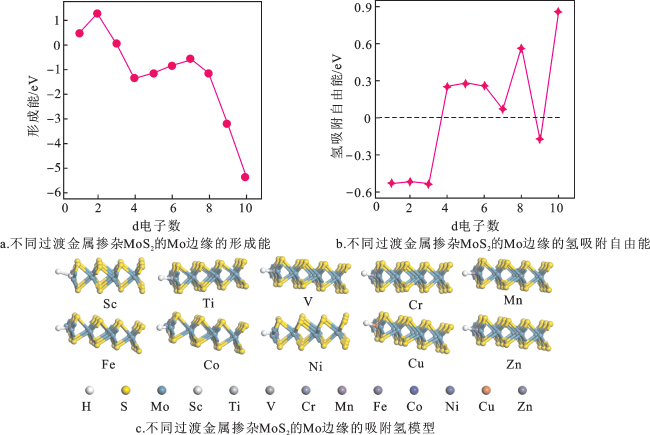
综上,相比于本征MoS2的Mo边缘( =-0.65 eV),Zn掺杂MoS2的Mo边缘的析氢活性变差( =0.86 eV),而其他的过渡金属掺杂MoS2的Mo边缘的析氢活性(|Δ |<0.65 eV)都有所提升;其中,Co掺杂MoS2的Mo边缘的Δ (0.07 eV)更靠近0 eV,说明在过渡金属掺杂MoS2的Mo边缘结构中,Co掺杂MoS2具有最佳的析氢活性。
图6a为不同过渡金属掺杂MoS2的S边缘的形成能。Ti掺杂MoS2的S边缘的Ef为正值(Ef/Ti=0.68 eV),其他元素掺杂MoS2的S边缘的Ef都为负值,说明除Ti以外的过渡金属元素掺杂MoS2的S边缘结构更易制备。图6b为不同过渡金属掺杂MoS2后S边缘的氢吸附自由能。Co掺杂MoS2的S边缘的 (0.07 eV)更接近0 eV,说明Co掺杂MoS2的S边缘结构具有更好的析氢活性。图6c为不同过渡金属掺杂MoS2的S边缘的吸附氢模型。不同过渡金属掺杂MoS2的S边缘后,MoS2的结构未发生明显变化,但H的吸附发生了变化;其中,Co掺杂MoS2后S边缘的S-H的键长为0.141 nm,而Cu掺杂MoS2后S边缘的S-H的键长为0.157 nm,说明不同过渡金属掺杂对MoS2的S边缘析氢活性有影响。
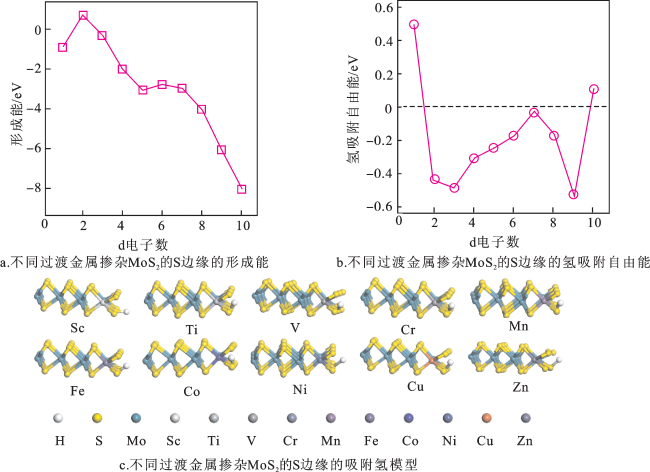
相比于本征MoS2的S边缘( =-0.46 eV),Sc、V和Cu掺杂MoS2的S边缘的析氢活性变差(Δ (Sc)=0.50 eV,Δ (V)=-0.48 eV,Δ (Cu)=0.54 eV),而其他的过渡金属掺杂MoS2的S边缘的析氢活性(|Δ |<0.46 eV)都有所提升;其中,Co掺杂MoS2的S边缘的Δ 为-0.03 eV更靠近0 eV,说明在过渡金属掺杂MoS2的S边缘结构中,Co掺杂MoS2具有最佳的析氢活性。
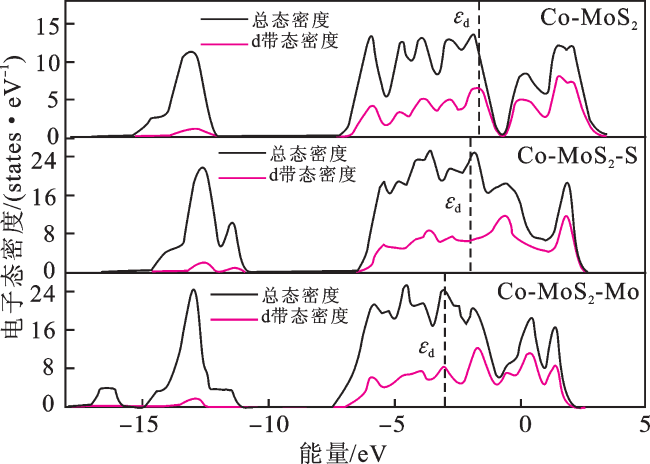
图7为Co分别掺杂在MoS2平面内,MoS2-Mo边缘,MoS2-S边缘3种体系的电子态密度(density of electronic states,DOS)。可以发现:DOS跨过费米能级,表明3种体系具有类似金属的导电性。在文献[29]中,d带态密度在H吸附过程中起着重要作用。例如,d带中心靠近费米能级,意味着反应中间体与它们的结合强度有改变,使得硫化物在H吸附步骤中表现出较快的反应速率[30]。Co掺杂在MoS2平面的d带中心(-1.70 eV)大于掺杂在MoS2-S边缘(-2.01 eV)和MoS2-Mo边缘(-3.04 eV)。表明Co掺杂导致MoS2的d带中心靠近费米能级,调控了H吸附的强度,从而提高了析氢反应活性。
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
3 结论
基于密度泛函理论的第一性原理计算,研究了不同过渡金属元素(Sc、Ti、V、Cr、Mn、Fe、Co、Ni、Cu、Zn)掺杂对MoS2平面、S边缘、Mo边缘的结构稳定性及析氢性能的影响。研究结果表明,本征MoS2的平面析氢性能远低于边缘析氢性能,而S边缘的析氢性能高于Mo边缘;无论表面吸附过渡金属原子还是掺杂均提高了MoS2平面的析氢性能;所考虑元素的掺杂对S边缘和Mo边缘析氢性能的影响不一;其中,Co掺杂MoS2的平面、S边缘及Mo边缘具有最佳的析氢性能,氢吸附的自由能变化分别为0.01 eV、-0.03 eV和0.07 eV。因此通过Co掺杂MoS2可以制备出高效的析氢催化剂。本工作不仅证实了掺杂可以提高MoS2的析氢活性,而且为MoS2基电催化剂的设计提供了理论基础。